Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chen, C.; Zhang, J. Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics. Encyclopedia. Available online: https://encyclopedia.pub/entry/56436 (accessed on 05 July 2026).
Chen C, Zhang J. Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics. Encyclopedia. Available at: https://encyclopedia.pub/entry/56436. Accessed July 05, 2026.
Chen, Can, Ji Zhang. "Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics" Encyclopedia, https://encyclopedia.pub/entry/56436 (accessed July 05, 2026).
Chen, C., & Zhang, J. (2024, March 20). Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics. In Encyclopedia. https://encyclopedia.pub/entry/56436
Chen, Can and Ji Zhang. "Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics." Encyclopedia. Web. 20 March, 2024.
Copy Citation
Glutamine, one of the most abundant amino acids in the body, plays diverse roles in cellular metabolism, ranging from bioenergetics to the synthesis of nucleotides, glutathione (GSH), and various other non-essential amino acids.
leukemia
amino acid starvation
combination therapy
1. Glutamine Catabolism and Its Regulation
Glutamine, one of the most abundant amino acids in the body [1], plays diverse roles in cellular metabolism, ranging from bioenergetics to the synthesis of nucleotides, glutathione (GSH), and various other non-essential amino acids [2]. Classified as a non-essential amino acid by itself, it can be synthesized de novo by using other carbon- and nitrogen-containing nutrients as precursors in mammalian cells [2]. It is also transported across cell and mitochondrial membranes by specific solute carrier family members, including SLC1A5, SLC38A2, SLC38A1, and SLC1A1 [3]. Its export occurs through the SLC7A5 antiporter in exchange for extracellular leucine [4].
The catabolism of glutamine initiates with its conversion to glutamate, which can be catalyzed by glutaminase (GLS) in many cancer cells [5][6], a process that also releases a free amide nitrogen molecule to biosynthetic pathways. Humans possess two types of glutaminase: kidney-type GLS1, distributed ubiquitously, and liver-type GLS2, primarily expressed in the liver [7]. In mammalian cells, the GLS1 gene encodes two isoforms of glutaminase: kidney-type glutaminase (KGA) and glutaminase C (GAC). Notably, the GAC isoform is predominantly found in acute myeloid leukemia (AML) [8]. Glutamate is subsequently converted by glutamate dehydrogenase (GDH) or by transaminases into α-ketoglutarate (α-KG). α-KG then can enter the tricarboxylic acid cycle (TCA), providing both energy and intermediates for macromolecule biosynthesis [9]. Remarkably, in certain cell types, glutamine serves as an equivalent energy source for ATP production as glucose [10]. Furthermore, glutamine is a precursor for nucleotides, is involved in the synthesis of other non-essential amino acids, and contributes to the formation of glutathione, a key cellular reducing equivalent to mitigate oxidative stress [11]. Indeed, many AML subtypes rely on producing glutathione to maintain intracellular redox balance [12], particularly in leukemia stem cells and residual leukemia cells after chemotherapy, suggesting vulnerability in redox homeostasis in these cells, which should be targeted to prevent leukemia recurrence or refractory disease [13].
Hematological malignancies heavily rely on glutamine for mitochondrial oxidative phosphorylation (OXPHOS), a vital energetic process to support cell survival and proliferation [8]. In leukemia, elevated expression of SLC1A5 facilitates glutamine uptake, a necessary process for the synthesis of glutathione and pyrimidine to drive chemo-resistance in the leukemic blast cells [14]. In an independent study, it was shown that the elevated expression of SLC1A5 is driven by the signal transducer and activator of transcription 3 (STAT3)–MYC protooncogene axis in AML stem and progenitor cells (LSCs), enhancing glutamine influx to support the TCA cycle and glutathione production [15].
2. Targeting Glutamine Metabolism in AML
Current strategies targeting glutamine metabolism in cancer cells focus on its depletion, glutaminase inhibition, and inhibition of glutamine uptake by blocking its membrane transporters. As a result, combined treatment strategies have emerged to combat drug resistance and enhance anti-tumor efficacy. AML cells are generally reliant on glutamine and mitochondrial OXPHOS for survival [8]. Notably, inhibitors of GLS1, such as BPTES, compound 938, and compound CB-839, are available, with CB-839 undergoing human clinical trials for hematological malignancies. Inhibiting glutamine metabolism via CB-839 significantly disrupts the production of glutathione in various AML types, leading to an accumulation of mitochondrial reactive oxygen species (mitoROS) and apoptotic cell death [8]. The inhibition of GLS1 induces apoptosis through caspase activation, predominantly involving the intrinsic mitochondrial apoptotic pathway [8]. In primary AML leukemic stem cells, high-level expression of BCL-2 supports OXPHOS, rendering the cells to be highly sensitive to BCL-2-specific inhibitors [16]. Combining CB-839 with one of these BCL-2 inhibitors, Venetoclax, demonstrates promising clinical potential for treating AML by inhibiting glutaminolysis, triggering mitochondrial depolarization, and inducing intrinsic caspase-dependent apoptosis, thus exhibiting potent anti-leukemic activity [8].
Combining glutaminase inhibition with agents that disrupt mitochondrial redox balance intensifies leukemic cells’ vulnerability to cell death. In a pre-clinical study, combining CB-839 with arsenic trioxide (ATO) or homoharringtonine (HHT), two known ROS inducers, significantly exacerbated cellular ROS production and cell death in AML cell lines in vitro and a secondary mouse AML model in vivo [17]. Furthermore, this combination therapy effectively eliminated primary AML colony-forming cells, indicating potential efficacy against leukemia stem cells. Notably, the CB-839 and HHT combination also demonstrates efficacy in treating acute lymphoblastic leukemia (ALL), another type of leukemia originating from lymphoid progenitor cells.
Mutations in the FLT3 receptor type-III kinase are prevalent in about 30% of AML patients, primarily characterized by FLT3 internal tandem duplication (ITD) mutations. These mutations represent an increased rate of relapse post-standard therapies and a poor prognosis [18]. Investigation by metabolomics and gene-expression profiling analyses revealed that in FLT3-ITD, AMLs specifically depend on glutamine metabolism during treatment targeting the tyrosine kinase activities of the FLT3 receptors. Inhibition of FLT3-ITD in these AMLs disrupts glutamine metabolism by blocking its uptake via an undefined mechanism. As a result, inhibition of glutamine uptake depletes glutathione and induces apoptosis due to increased mitochondrial oxidative stress [19][20]. Combining AC220, a FLT3 inhibitor, with CB-839 further depletes glutathione, induces mitochondrial ROS, and triggers apoptotic cell death synergistically [20][21]. In vivo experiments using CB-839 demonstrated enhanced elimination of leukemic cells when AC220 was given simultaneously, significantly improving survival in a patient-derived xenograft AML model [20]. Similarly, the combination of imatinib, a BCR/ABL inhibitor, with CB-839 amplifies apoptosis in BCR-ABL-positive cells, characterized by increased intracellular ROS production and reduced oxygen consumption [21]. The completed clinical trial (Table 1), registered as NCT02071927, utilized a combination of CB-839 and azacytidine, a DNA methyltransferase (DNMT) inhibitor known for its demethylating effects in vitro and in vivo. Until now, no results from this trial have been made public. However, a similar combination has demonstrated efficacy in patients with myelodysplastic syndromes, as evidenced by the study registered as NCT03047993 (Table 1). In this trial, an objective response, defined as complete remission (CR), morphologic CR, or hematologic improvement, was achieved in 15 out of 23 patients, accounting for 65.2% of the participants [22].
Table 1. Ongoing and completed clinical trials involving perturbation of amino acid metabolism.
| Registered Number | Disease | Phase | Group | Status | Reference |
|---|---|---|---|---|---|
| NCT02071927 | R/R leukemia | I | Single-agent CB-839/CB-839 and AZA | Complete | N.A |
| NCT03047993 | Advanced MDS | I/II | CB-839 and AZA | Complete | [22] |
| NCT04666649 | R/R AML | I | Ven-PegC | Ongoing | N.A |
| NCT02899286 | R/R AML | II | PEG-BCT-100 | Complete | N.A |
| NCT01551628 | R/R leukemia | I | Recombinant human arginase1 Peg-5000 | Terminated(slow patient recruitment) | N.A |
| NCT02732184 | R/R AML or MDS | II | Co-ArgI-PEG modified human arginase I | Complete | N.A |
| NCT01910012 | R/RAML | II | ADI-PEG 20 | Complete | [1] |
| NCT05001828 | High risk AML | I | ADI-PEG 20, Venetoclax and Azacitidine | Ongoing | N.A |
| NCT02835729 | ND-AML | I | Indoximodin, Idarubicin and Cytarabine | Complete | [23] |
| 2006-005694-21 (EWALL-PH-01) |
ND-Ph+ and/or CR-ABL1+ ALL | II | Dasatinib, cytarabine, asparaginase and methotrexate | Complete | [24] |
| NCT01085617(UKALL14) | newly diagnosed ALL | III | PEG-ASP and standard induction regimen (ph + disease received continuous oral imatinib) | Complete | [25] |
3. Combination Therapeutics Involving Glutamine Perturbation and Traditional Chemo-Agents
There is no drug that can deplete glutamine in circulation specifically till today. However, L-asparaginase, a chemo-agent that is used in ALL patients via degrading asparagine to aspartate and ammonia has caught attention because L-asparaginase can also degrade glutamine but as a less effective substrate [26]. Indeed, a clinical study using crisantaspase, an asparaginase purified from Erwinia chrysanthemi, showed responses in relapsed AML patients, which correlated with its capacity to deplete glutamine in the plasma of these patients [27]. Pegcrisantaspase (PegC), a long-acting form of crisantaspase, effectively depletes glutamine and asparagine, and inhibits mRNA translation and cellular protein synthesis, leading to cell death in leukemic cells in vitro [28]. The combination of Venetoclax, a BCL2 inhibitor, with PegC enhanced the interaction between eIF4E and 4EBP1 within the cap-binding complex to inhibit global mRNA translation, which reduced the expression of MCL-1 protein and caused cell death in vitro and synergistically inhibited leukemia progression in a PDX AML model in vivo. In this study, it was shown that glutamine depletion by PegC played a major role in this process [28]. A clinical trial focusing on relapsed/refractory acute myeloid leukemia (R/R AML) is currently ongoing, investigating the combination of PegC and Venetoclax (NCT04666649, Table 1).
Residual AML cells exhibit transient metabolic adaptation that contributes to their chemo-resistance. Studies in mouse AML models suggest that strategically manipulating specific metabolic pathways, such as glutamine catabolism and pyrimidine synthesis, holds therapeutic promise [14]. Glutamine analogs and antagonists, such as 6-diazo-5-oxo-L-norleucine (DON), inhibit a spectrum of enzymes that utilize glutamine as substrates [29]. Administering DON concurrently with chemotherapy (CT) showed no difference in survival compared to CT alone. However, when DON treatment followed CT and covered the phase of maximal response, a distinct survival benefit was observed. Short-term DON treatment post-CT notably enhanced AML cell elimination, evident by increased apoptotic cells and higher instances of double-stranded DNA breaks in residual cells [14]. While DON was tested in clinical trials for both solid tumors and leukemia since the 1980s, it was however abandoned due to its limited efficacy as a single agent and the occurrence of dose-dependent side effects [29].
Targeting glutamine metabolism in leukemia aims to reduce its uptake, block its catabolism, and thereby prevent its further utilization (Figure 1). In recent work, a non-specific inhibitor of glutamine transporter SLC1A5, namely L-c-glutamyl-p-nitroanilide (GPNA), showed efficacy in inhibiting leukemia progression in a xenograft AML model in vivo [30]. It will be interesting in the future to test more specific inhibitors, such as V-9302, which has been found to be effective in solid tumor models [31]. Notably, inhibitors targeting FLT3-ITD show promise in inhibiting glutamine uptake and exhibiting synergistic effects with glutaminase inhibitors. Given the importance of GSH, a vital antioxidant produced via glutamine catabolism, in mitigating oxidative-stress-induced cell death, combining glutamine inhibition with other chemotherapy drugs that induce ROS seems to be a rational choice. Glutamine deprivation might overcome resistance to BCL-2 inhibitors by perturbing mitochondrial integrity and function. In the future, it warrants further investigation of other glutamine-dependent biosynthetic pathways, such as nucleotide biosynthesis, to determine its potential synergy with other chemotherapeutics in leukemia.
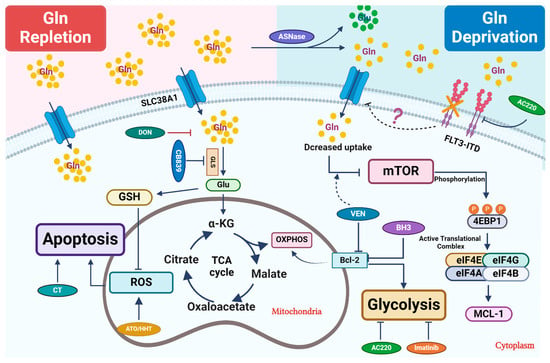
Figure 1. Therapeutic strategies that perturb glutamine metabolism. Glutamine (Gln) is transported into cells through cell surface transporters (SLC38A1). It is used as a precursor for protein synthesis. Intracellularly, glutamine is broken down into glutamate (Glu) by enzymes like GLS. Glutamate enters the TCA cycle via further catabolism into a-ketoglutarate (α-KG), which is critical to replenish the TCA cycle intermediates for other biosynthetic pathways or ATP production through oxidative phosphorylation (OXPHOS). Glu is also a critical precursor to synthesize glutathione (GSH), a key antioxidant to mitigate oxidative stress in leukemic cells. Combined treatment primarily focuses on blocking glutamine uptake or blocking its conversion into glutamate. In certain settings, L-asparaginase (ASNase) can deplete extracellular glutamine, which further reduces its intracellular utilization. Venetoclax (VEN) enhances the inhibitory effect of amino acid deprivation caused by ASNase treatment on mTOR, ultimately leading to the suppression of MCL-1 protein expression. 6-diazo-5-oxo-L-norleucine, also known as DON, is a glutamine analog. It disrupts multiple metabolic reactions that utilize glutamine as a substrate. CB-839, a GLS inhibitor, inhibits glutamate production and significantly increases ROS when combined with oxidizing agents, such as ATO/HHT. BCL-2 enhances mitochondrial OXPHOS for energy production, and thus its inhibition synergizes with CB-839 to disrupt energy production and mitochondrial function. Additionally, FLT3-TKI (AC220) reduces glutamine uptake and utilization in leukemic cells and thus shows synergy with CB-839, likely through perturbing bioenergetics.
References
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634.
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315.
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 2014, 15, 169–184.
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutte, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887.
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765.
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740.
- Nguyen, T.T.; Katt, W.P.; Cerione, R.A. Alone and together: Current approaches to targeting glutaminase enzymes as part of anti-cancer therapies. Future Drug. Discov. 2023, 4, FDD79.
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356.
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787.
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712.
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684.
- Goto, M.; Miwa, H.; Shikami, M. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig. 2014, 32, 241–247.
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558.
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.e396.
- Amaya, M.; Inguva, A.; Pei, S. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood 2021, 139, 584–596.
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341.
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090.
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312.
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678.
- Gregory, M.A.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58.
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653.
- Saxena, K.; Konopleva, M.; Bhagat, T.D.; Guerra, V.A.; Maduike, R.; Tiziani, S.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Kadia, T.M.; et al. AZA + Glutaminase Inhibition with Telaglenastat (CB-839) for Advanced MDS: An Updated Interim Analysis. Blood 2020, 136, 31–32.
- Emadi, A.; Duong, V.H.; Pantin, J.; Imran, M.; Koka, R.; Singh, Z.; Sausville, E.A.; Law, J.Y.; Lee, S.T.; Shi, H.; et al. Indoximod Combined with Standard Induction Chemotherapy Is Well Tolerated and Induces a High Rate of Complete Remission with MRD-Negativity in Patients with Newly Diagnosed AML: Results from a Phase 1 Trial. Blood 2018, 132, 332.
- Rousselot, P.; Coude, M.M.; Gokbuget, N.; Gambacorti Passerini, C.; Hayette, S.; Cayuela, J.M.; Huguet, F.; Leguay, T.; Chevallier, P.; Salanoubat, C.; et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood 2016, 128, 774–782.
- Patel, B.; Kirkwood, A.A.; Dey, A.; Marks, D.I.; McMillan, A.K.; Menne, T.F.; Micklewright, L.; Patrick, P.; Purnell, S.; Rowntree, C.J.; et al. Pegylated-asparaginase during induction therapy for adult acute lymphoblastic leukaemia: Toxicity data from the UKALL14 trial. Leukemia 2017, 31, 58–64.
- Avramis, V.I. Asparaginases: Biochemical pharmacology and modes of drug resistance. Anticancer. Res. 2012, 32, 2423–2437.
- Emadi, A.; Law, J.Y.; Strovel, E.T.; Lapidus, R.G.; Jeng, L.J.B.; Lee, M.; Blitzer, M.G.; Carter-Cooper, B.A.; Sewell, D.; Van Der Merwe, I.; et al. Asparaginase Erwinia chrysanthemi effectively depletes plasma glutamine in adult patients with relapsed/refractory acute myeloid leukemia. Cancer Chemother. Pharmacol. 2018, 81, 217–222.
- Emadi, A.; Kapadia, B.; Bollino, D.; Bhandary, B.; Baer, M.R.; Niyongere, S.; Strovel, E.T.; Kaizer, H.; Chang, E.; Choi, E.Y.; et al. Venetoclax and pegcrisantaspase for complex karyotype acute myeloid leukemia. Leukemia 2021, 35, 1907–1924.
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832.
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403.
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
560
Revisions:
2 times
(View History)
Update Date:
21 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No