 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Asher Ornoy | -- | 6353 | 2024-03-14 07:31:53 | | | |
| 2 | Lindsay Dong | Meta information modification | 6353 | 2024-04-15 07:19:34 | | |
Video Upload Options
Valproic acid (VPA) is a very effective anticonvulsant and mood stabilizer with relatively few side effects. Being an epigenetic modulator, it undergoes clinical trials for the treatment of advanced prostatic and breast cancer. However, in pregnancy, it seems to be the most teratogenic antiepileptic drug. Among the proven effects are congenital malformations in about 10%. The more common congenital malformations are neural tube defects, cardiac anomalies, urogenital malformations including hypospadias, skeletal malformations and orofacial clefts. These effects are dose related; daily doses below 600 mg have a limited teratogenic potential. VPA, when added to other anti-seizure medications, increases the malformations rate. It induces malformations even when taken for indications other than epilepsy, adding to the data that epilepsy is not responsible for the teratogenic effects.
1. Introduction
2. The Therapeutic Effects of VPA
3. Possible Damage and Major Congenital Malformations (MCMs) Caused by VPA in Pregnancy
4. VPA and Dose-Related MCMs
5. VPA and Polytherapy
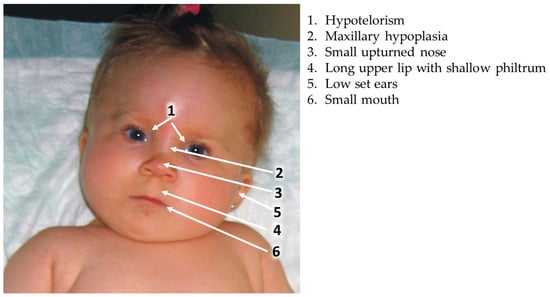
6. Valproic Acid Syndrome (Valproic Acid Spectrum Disorder)
7. VPA and Neurodevelopmental Problems
7.1. Impaired Neurodevelopment
7.2. Intellectual and Learning Disabilities
7.3. VPA and ADHD
7.4. VPA and Autism Spectrum Disorder (ASD)
8. VPA and Folic Acid Administration in Human
9. VPA Transplacental Passage and Secretion into Milk and Semen
10. Malformations in Children of Untreated Epileptic Women
11. Animal Studies on VPA-Induced Teratogenicity
11.1. VPA-Induced MCMs–General
11.2. VPA-Induced General (Microscopic) Effects on the Nervous Tissue
11.3. VPA-Induced Microscopic Pathological Changes in the Liver and Heart
11.4. VPA-Induced Congenital Malformations in Animals
12. VPA-Induced Autistic-like Behavior in Animals
13. VPA-Induced Changes in Gene Expression in the Brain of Animals
14. The Suggested Mechanism of the Teratogenic Action of VPA
14.1. Folate One-Carbon Metabolism (OCM) and Folate Deficiency
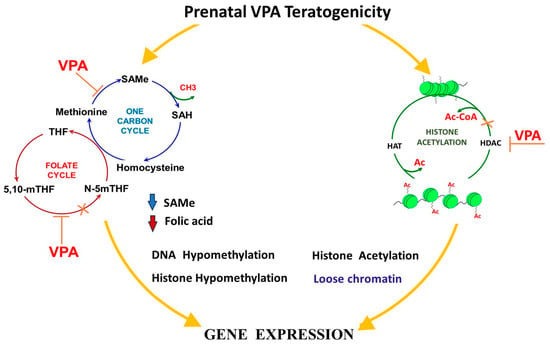
14.2. Alterations in the SAMe Cycle and VPA-Induced Malformations
14.3. The Inhibition of Histone Deacetylases (HDAC)
14.4. Increased Oxidative Stress
14.5. Mitochondrial Dysfunction
14.6. Inositol Depletion
15. The Prevention of VPA Teratogenicity in Animals in Relation to Congenital Malformations
15.1. Attempts to Minimize VPA Teratogenicity
15.2. VPA and Folic Acid (FA) Administration in Animals
16. The Prevention of the VPA Induction of ASD in Preclinical Animal Models
17. Conclusions
References
- Ornoy, A. The impact of intrauterine exposure versus postnatal environment in neurodevelopmental toxicity: Long-term neurobehavioral studies in children at risk for developmental disorders. Toxicol. Lett. 2003, 140–141, 171–181.
- Arnon, J.; Shechtman, S.; Ornoy, A. The use of psychiatric drugs in pregnancy and lactation. Isr. J. Psychiatry Relat. Sci. 2000, 37, 205–222.
- Jentink, J.; Loane, M.A.; Dolk, H.; Barisic, I.; Garne, E.; Morris, J.K.; de Jong-van den Berg, L.T. Valproic acid monotherapy in pregnancy and major congenital malformations. New Engl. J. Med. 2010, 362, 2185–2193.
- Ornoy, A. Neuroteratogens in man: An overview with special emphasis on the teratogenicity of antiepileptic drugs in pregnancy. Reprod. Toxicol. 2006, 22, 214–226.
- Kultima, K.; Nyström, A.-M.; Scholz, B.; Gustafson, A.-L.; Dencker, L.; Stigson, M. Valproic Acid Teratogenicity: A Toxicogenomics Approach. Environ. Health Perspect. 2004, 112, 1225–1235.
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10.
- Diav-Citrin, O.; Shechtman, S.; Bar-Oz, B.; Cantrell, D.; Arnon, J.; Ornoy, A. Pregnancy outcome after in utero exposure to valproate: Evidence of dose relationship in teratogenic effect. CNS Drugs 2008, 22, 325–334.
- Morrow, J.; Russell, A.; Guthrie, E.; Parsons, L.; Robertson, I.; Waddell, R.; Irwin, B.; McGivern, R.C.; Morrison, P.J.; Craig, J. Malformation risks of antiepileptic drugs in pregnancy: A prospective study from the UK Epilepsy and Pregnancy Register. J. Neurol. Neurosurg. Psychiatry 2006, 77, 193–198.
- Bromley, R.; Weston, J.; Adab, N.; Greenhalgh, J.; Sanniti, A.; McKay, A.J. Treatment for epilepsy in pregnancy: Neurodevelopmental outcomes in the child. Cochrane Database Syst. Rev. 2014, 10, CD010236.
- Weston, J.; Bromley, R.; Jackson, C.F.; Adab, N.; Clayton-Smith, J.; Greenhalgh, J.; Hounsome, J.; McKay, A.J.; Tudur Smith, C.; Marson, A.G. Monotherapy treatment of epilepsy in pregnancy: Congenital malformation outcomes in the child. Cochrane Database Syst. Rev. 2016, 11, CD010224.
- Kini, U.; Adab, N.; Vinten, J.; Fryer, A.; Clayton-Smith, J. Dysmorphic features: An important clue to the diagnosis and severity of fetal anticonvulsant syndromes. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F90–F95.
- Fried, S.; Kozer, E.; Nulman, I.; Einarson, T.R.; Koren, G. Malformation rates in children of women with untreated epilepsy: A meta-analysis. Drug Saf. 2004, 27, 197–202.
- Anmella, G.; Pacchiarotti, I.; Cubała, W.J.; Dudek, D.; Maina, G.; Thomas, P.; Vieta, E. Expert advice on the management of valproate in women with bipolar disorder at childbearing age. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2019, 29, 1199–1212.
- Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. Int. J. Mol. Sci. 2020, 21, 3721.
- Singh, D.; Gupta, S.; Verma, I.; Morsy, M.A.; Nair, A.B.; Ahmed, A.F. Hidden pharmacological activities of valproic acid: A new insight. Biomed. Pharmacother. 2021, 142, 112021.
- Ornoy, A.; Arnon, J. Clinical teratology. West. J. Med. 1993, 159, 382.
- Philbrook, N.A.; Nikolovska, A.; Maciver, R.D.; Belanger, C.L.; Winn, L.M. Characterizing the effects of in utero exposure to valproic acid on murine fetal heart development. Birth Defects Res. 2019, 111, 1551–1560.
- Bromley, R.; Adab, N.; Bluett-Duncan, M.; Clayton-Smith, J.; Christensen, J.; Edwards, K.; Greenhalgh, J.; Hill, R.A.; Jackson, C.F.; Khanom, S.; et al. Monotherapy treatment of epilepsy in pregnancy: Congenital malformation outcomes in the child. Cochrane Database Syst. Rev. 2023, 8, CD010224.
- Yerby, M.S. Pregnancy, teratogenesis, and epilepsy. Neurol. Clin. 1994, 12, 749–771.
- Bjerkedal, T.; Czeizel, A.; Goujard, J.; Kallen, B.; Mastroiacova, P.; Nevin, N.; Oakley, G., Jr.; Robert, E. Valproic acid and spina bifida. Lancet 1982, 2, 1096.
- Verloes, A.; Frikiche, A.; Gremillet, C.; Paquay, T.; Decortis, T.; Rigo, J.; Senterre, J. Proximal phocomelia and radial ray aplasia in fetal valproic syndrome. Eur. J. Pediatr. 1990, 149, 266–267.
- Kim, H.H.; Laufer, M.R. Developmental abnormalities of the female reproductive tract. Curr. Opin. Obstet. Gynecol. 1994, 6, 518–525.
- Okada, A.; Kurihara, H.; Aoki, Y.; Bialer, M.; Fujiwara, M. Amidic modification of valproic acid reduces skeletal teratogenicity in mice. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2004, 71, 47–53.
- Yerby, M.S. Management issues for women with epilepsy: Neural tube defects and folic acid supplementation. Neurology 2003, 61, S23–S26.
- Koren, G.; Nava-Ocampo, A.A.; Moretti, M.E.; Sussman, R.; Nulman, I. Major malformations with valproic acid. Can. Fam. Physician Med. Fam. Can. 2006, 52, 441–442, 444, 447.
- Perucca, E.; Tomson, T. Prenatal exposure to antiepileptic drugs. Lancet 2006, 367, 1467–1469.
- Malm, H.; Kajantie, E.; Kivirikko, S.; Kääriäinen, H.; Peippo, M.; Somer, M. Valproate embryopathy in three sets of siblings: Further proof of hereditary susceptibility. Neurology 2002, 59, 630–633.
- Alsdorf, R.; Wyszynski, D.F. Teratogenicity of sodium valproate. Expert. Opin. Drug Saf. 2005, 4, 345–353.
- Rodríguez-Pinilla, E.; Mejías, C.; Prieto-Merino, D.; Fernández, P.; Martínez-Frías, M.L. Risk of hypospadias in newborn infants exposed to valproic acid during the first trimester of pregnancy: A case-control study in Spain. Drug Saf. 2008, 31, 537–543.
- Eadie, M.J.; Vajda, F.J. Should valproate be taken during pregnancy? Ther. Clin. Risk Manag. 2005, 1, 21–26.
- Campbell, E.; Kennedy, F.; Russell, A.; Smithson, W.H.; Parsons, L.; Morrison, P.J.; Liggan, B.; Irwin, B.; Delanty, N.; Hunt, S.J.; et al. Malformation risks of antiepileptic drug monotherapies in pregnancy: Updated results from the UK and Ireland Epilepsy and Pregnancy Registers. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1029–1034.
- Artama, M.; Auvinen, A.; Raudaskoski, T.; Isojärvi, I.; Isojärvi, J. Antiepileptic drug use of women with epilepsy and congenital malformations in offspring. Neurology 2005, 64, 1874–1878.
- Kaneko, S.; Battino, D.; Andermann, E.; Wada, K.; Kan, R.; Takeda, A.; Nakane, Y.; Ogawa, Y.; Avanzini, G.; Fumarola, C.; et al. Congenital malformations due to antiepileptic drugs. Epilepsy Res. 1999, 33, 145–158.
- Matalon, S.; Schechtman, S.; Goldzweig, G.; Ornoy, A. The teratogenic effect of carbamazepine: A meta-analysis of 1255 exposures. Reprod. Toxicol. 2002, 16, 9–17.
- GSK The Lamotrigine Pregnancy Registry. Available online: http://pregnancyregistry.gsk.com/documents/lamreport (accessed on 1 January 2008).
- Laegreid, L.; Kyllerman, M.; Hedner, T.; Hagberg, B.; Viggedahl, G. Benzodiazepine amplification of valproate teratogenic effects in children of mothers with absence epilepsy. Neuropediatrics 1993, 24, 88–92.
- Holmes, L.B.; Mittendorf, R.; Shen, A.; Smith, C.R.; Hernandez-Diaz, S. Fetal effects of anticonvulsant polytherapies: Different risks from different drug combinations. Arch Neurol. 2011, 68, 1275–1281.
- DiLiberti, J.H.; Farndon, P.A.; Dennis, N.R.; Curry, C.J. The fetal valproate syndrome. Am. J. Med. Genet. 1984, 19, 473–481.
- Kozma, C. Valproic acid embryopathy: Report of two siblings with further expansion of the phenotypic abnormalities and a review of the literature. Am. J. Med. Genet. 2001, 98, 168–175.
- Ardinger, H.H.; Atkin, J.F.; Blackston, R.D.; Elsas, L.J.; Clarren, S.K.; Livingstone, S.; Flannery, D.B.; Pellock, J.M.; Harrod, M.J.; Lammer, E.J.; et al. Verification of the fetal valproate syndrome phenotype. Am. J. Med. Genet. 1988, 29, 171–185.
- Mawer, G.; Clayton-Smith, J.; Coyle, H.; Kini, U. Outcome of pregnancy in women attending an outpatient epilepsy clinic: Adverse features associated with higher doses of sodium valproate. Seizure 2002, 11, 512–518.
- Moore, S.J.; Turnpenny, P.; Quinn, A.; Glover, S.; Lloyd, D.J.; Montgomery, T.; Dean, J.C. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 2000, 37, 489–497.
- Winter, R.M.; Donnai, D.; Burn, J.; Tucker, S.M. Fetal valproate syndrome: Is there a recognisable phenotype? J. Med. Genet. 1987, 24, 692–695.
- Dean, J.C.; Moore, S.J.; Turnpenny, P.D. Developing diagnostic criteria for the fetal anticonvulsant syndromes. Seizure 2000, 9, 233–234.
- Rihtman, T.; Parush, S.; Ornoy, A. Developmental outcomes at preschool age after fetal exposure to valproic acid and lamotrigine: Cognitive, motor, sensory and behavioral function. Reprod. Toxicol. 2013, 41, 115–125.
- Koch, S.; Jäger-Roman, E.; Lösche, G.; Nau, H.; Rating, D.; Helge, H. Antiepileptic drug treatment in pregnancy: Drug side effect in the neonate and neurological outcome. Acta Paediatr. Int. J. Paediatr. 1996, 85, 739–746.
- Nicolai, J.; Vles, J.S.; Aldenkamp, A.P. Neurodevelopmental delay in children exposed to antiepileptic drugs in utero: A critical review directed at structural study-bias. J. Neurol. Sci. 2008, 271, 1–14.
- Christianson, A.L.; Chester, N.; Kromberg, J.G.R. Fetal Valproate Syndrome: Clinical and Neuro-developmental Features in Two Sibling Pairs. Dev. Med. Child Neurol. 1994, 36, 361–369.
- Dean, J.C.; Hailey, H.; Moore, S.J.; Lloyd, D.J.; Turnpenny, P.D.; Little, J. Long term health and neurodevelopment in children exposed to antiepileptic drugs before birth. J. Med. Genet. 2002, 39, 251–259.
- Viinikainen, K.; Eriksson, K.; Mönkkönen, A.; Aikiä, M.; Nieminen, P.; Heinonen, S.; Kälviäinen, R. The effects of valproate exposure in utero on behavior and the need for educational support in school-aged children. Epilepsy Behav. 2006, 9, 636–640.
- Adab, N.; Jacoby, A.; Smith, D.; Chadwick, D. Additional educational needs in children born to mothers with epilepsy. J. Neurol. Neurosurg. Psychiatry 2001, 70, 15–21.
- Adab, N.; Kini, U.; Vinten, J.; Ayres, J.; Baker, G.; Clayton-Smith, J.; Coyle, H.; Fryer, A.; Gorry, J.; Gregg, J.; et al. The longer term outcome of children born to mothers with epilepsy. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1575–1583.
- Langer, B.; Haddad, J.; Gasser, B.; Maubert, M.; Schlaeder, G. Isolated fetal bilateral radial ray reduction associated with valproic acid usage. Fetal Diagn. Ther. 1994, 9, 155–158.
- Gaily, E.; Kantola-Sorsa, E.; Hiilesmaa, V.; Isoaho, M.; Matila, R.; Kotila, M.; Nylund, T.; Bardy, A.; Kaaja, E.; Granström, M.L. Normal intelligence in children with prenatal exposure to carbamazepine. Neurology 2004, 62, 28–32.
- Eriksson, K.; Viinikainen, K.; Mönkkönen, A.; Äikiä, M.; Nieminen, P.; Heinonen, S.; Kälviäinen, R. Children exposed to valproate in utero—population based evaluation of risks and confounding factors for long-term neurocognitive development. Epilepsy Res. 2005, 65, 189–200.
- Shallcross, R.; Bromley, R.L.; Irwin, B.; Bonnett, L.J.; Morrow, J.; Baker, G.A. Child development following in utero exposure: Levetiracetam vs sodium valproate. Neurology 2011, 76, 383–389.
- Conners, C.K. Conner’s Rating Scales—Revised Technical Manual; Multi-Health Systems Inc.: Toronto, ON, Canada, 1997.
- Cohen, M.J.; Meador, K.J.; May, R.; Loblein, H.; Conrad, T.; Baker, G.A.; Bromley, R.L.; Clayton-Smith, J.; Kalayjian, L.A.; Kanner, A.; et al. Fetal antiepileptic drug exposure and learning and memory functioning at 6 years of age: The NEAD prospective observational study. Epilepsy Behav. 2019, 92, 154–164.
- Williams, P.G.; Hersh, J.H. A male with fetal valproate syndrome and autism. Dev. Med. Child Neurol. 1997, 39, 632–634.
- Rasalam, A.D.; Hailey, H.; Williams, J.H.; Moore, S.J.; Turnpenny, P.D.; Lloyd, D.J.; Dean, J.C. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol. 2005, 47, 551–555.
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703.
- Wiggs, K.K.; Rickert, M.E.; Sujan, A.C.; Quinn, P.D.; Larsson, H.; Lichtenstein, P.; Oberg, A.S.; D’Onofrio, B.M. Antiseizure medication use during pregnancy and risk of ASD and ADHD in children. Neurology 2020, 95, e3232–e3240.
- Wood, A.G.; Nadebaum, C.; Anderson, V.; Reutens, D.; Barton, S.; O’Brien, T.J.; Vajda, F. Prospective assessment of autism traits in children exposed to antiepileptic drugs during pregnancy. Epilepsia 2015, 56, 1047–1055.
- Honybun, E.; Thwaites, R.; Malpas, C.B.; Rayner, G.; Anderson, A.; Graham, J.; Hitchcock, A.; O’Brien, T.J.; Vajda, F.J.E.; Perucca, P. Prenatal valproate exposure and adverse neurodevelopmental outcomes: Does sex matter? Epilepsia 2021, 62, 709–719.
- Petersen, I.; McCrea, R.L.; Sammon, C.J.; Osborn, D.P.J.; Evans, S.J.; Cowen, P.J.; Freemantle, N.; Nazareth, I. Risks and benefits of psychotropic medication in pregnancy: Cohort studies based on UK electronic primary care health records. Health Technol. Assess. 2016, 20, 1–176.
- Hisle-Gorman, E.; Susi, A.; Stokes, T.; Gorman, G.; Erdie-Lalena, C.; Nylund, C.M. Prenatal, perinatal, and neonatal risk factors of autism spectrum disorder. Pediatr. Res. 2018, 84, 190–198.
- Nau, H.; Schafer, H.; Rating, D.; Jakobs, C.; Helge, H. Placental transfer and neonatal pharmacokinetics of valproic acid and some of its metabolites. In Epilepsy, Pregnancy, and the Child; Janz, D., Bossi, L., Dam, M., Helge, H., Richens, A., Schmidt, D., Eds.; Raven Press: New York, NY, USA, 1982; pp. 367–372.
- Albani, F.; Riva, R.; Contin, M.; Baruzzi, A.; Altomare, M.; Merlini, G.P.; Perucca, E. Differential transplacental binding of valproic acid: Influence of free fatty acids. Br. J. Clin. Pharmacol. 1984, 17, 759–762.
- Kacirova, I.; Grundmann, M.; Brozmanova, H. Serum levels of valproic acid during delivery in mothers and in umbilical cord—Correlation with birth length and weight. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2015, 159, 569–575.
- Kacirova, I.; Grundmann, M.; Brozmanova, H. Valproic Acid Concentrations in Mothers, Colostrum and Breastfed Infants during the Early Postpartum Period: Comparison with Concentrations Determined during Delivery and in the Mature Milk Period. Pharmaceutics 2021, 13, 2074.
- Von Unruh, G.E.; Froescher, W.; Hoffmann, F.; Niesen, M. Valproic acid in breast milk: How much is really there? Ther. Drug Monit. 1984, 6, 272–276.
- The WHO Working Group. Drugs and Human Lactation; Bennet, P.N., Ed.; Elsevier: Amsterdam, The Netherlands; New York, NY, USA; Oxford, UK, 1988; pp. 341–342.
- Shapiro, S.; Hartz, S.C.; Siskind, V.; Mitchell, A.A.; Slone, D.; Rosenberg, L.; Monson, R.R.; Heinonen, O.P. Anticonvulsants and parental epilepsy in the development of birth defects. Lancet 1976, 1, 272–275.
- Kaaja, E.; Kaaja, R.; Hiilesmaa, V. Major malformations in offspring of women with epilepsy. Neurology 2003, 60, 575–579.
- Bold, J.; Sakata-Haga, H.; Fukui, Y. Spinal nerve defects in mouse embryos prenatally exposed to valproic acid. Anat. Sci. Int. 2018, 93, 35–41.
- Di Renzo, F.; Giavini, E.; Menegola, E. Methionine pretreatment enhances the effects of valproate on axial development in a cd1 mouse model. Birth Defects Res. Part B-Dev. Reprod. Toxicol. 2013, 98, 328–333.
- Shafique, S.; Winn, L.M. Gestational valproic acid exposure induces epigenetic modifications in murine decidua. Placenta 2021, 107, 31–40.
- Tiboni, G.M.; Ponzano, A. Prevention of valproic acid-induced neural tube defects by sildenafil citrate. Reprod. Toxicol. 2015, 56, 175–179.
- Elphick, L.M.; Pawolleck, N.; Guschina, I.A.; Chaieb, L.; Eikel, D.; Nau, H.; Harwood, J.L.; Plant, N.J.; Williams, R.S. Conserved valproic-acid-induced lipid droplet formation in Dictyostelium and human hepatocytes identifies structurally active compounds. Dis. Models Mech. 2012, 5, 231–240.
- Goetzl, L. Environmental agents and reproductive risk. In Queenan’s Management of High-Risk Pregnancy: An Evidence-Based Approach; Wiley-Blackwell: Hoboken, NJ, USA, 2012; pp. 32–40.
- Salimi, A.; Alyan, N.; Akbari, N.; Jamali, Z.; Pourahmad, J. Selenium and L-carnitine protects from valproic acid-Induced oxidative stress and mitochondrial damages in rat cortical neurons. Drug Chem. Toxicol. 2022, 45, 1150–1157.
- Shafique, S.; Winn, L.M. Role of Cbp, p300 and Akt in valproic acid induced neural tube defects in CD-1 mouse embryos. Reprod. Toxicol. 2020, 95, 86–94.
- Takla, T.N.; Luo, J.; Sudyk, R.; Huang, J.; Walker, J.C.; Vora, N.L.; Sexton, J.Z.; Parent, J.M.; Tidball, A.M. A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects. Cells 2023, 12, 1697.
- Tiboni, G.M.; Ponzano, A.; Ferrone, A.; Franceschelli, S.; Speranza, L.; Patruno, A. Valproic acid alters nitric oxide status in neurulating mouse embryos. Reprod. Toxicol. 2021, 99, 152–159.
- Wachholz, G.E.; Rengel, B.D.; Vargesson, N.; Fraga, L.R. From the Farm to the Lab: How Chicken Embryos Contribute to the Field of Teratology. Front. Genet. 2021, 12, 666726.
- Binkerd, P.E.; Rowland, J.M.; Nau, H.; Hendrickx, A.G. Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague-Dawley rats. Fundam. Appl. Toxicol. Off. J. Soc. Toxicol. 1988, 11, 485–493.
- Lin, Y.L.; Bialer, M.; Cabrera, R.M.; Finnell, R.H.; Wlodarczyk, B.J. Teratogenicity of valproic acid and its constitutional isomer, amide derivative valnoctamide in mice. Birth Defects Res. 2019, 111, 1013–1023.
- Hendrickx, A.G.; Nau, H.; Binkerd, P.; Rowland, J.M.; Rowland, J.R.; Cukierski, M.J.; Cukierski, M.A. Valproic acid developmental toxicity and pharmacokinetics in the rhesus monkey: An interspecies comparison. Teratology 1988, 38, 329–345.
- Jazayeri, D.; Braine, E.; McDonald, S.; Dworkin, S.; Powell, K.L.; Griggs, K.; Vajda, F.J.E.; O’Brien, T.J.; Jones, N.C. A rat model of valproate teratogenicity from chronic oral treatment during pregnancy. Epilepsia 2020, 61, 1291–1300.
- Menegola, E.; Broccia, M.L.; Nau, H.; Prati, M.; Ricolfi, R.; Giavini, E. Teratogenic effects of sodium valproate in mice and rats at midgestation and at term. Teratog. Carcinog. Mutagen. 1996, 16, 97–108.
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicol. Teratol. 2019, 71, 64–74.
- Mowery, T.M.; Wilson, S.M.; Kostylev, P.V.; Dina, B.; Buchholz, J.B.; Prieto, A.L.; Garraghty, P.E. Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way; Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way. Int. J. Dev. Neurosci. 2015, 40, 15–23.
- Hara, Y.; Maeda, Y.; Kataoka, S.; Ago, Y.; Takuma, K.; Matsuda, T. Effect of prenatal valproic acid exposure on cortical morphology in female mice. J. Pharmacol. Sci. 2012, 118, 543–546.
- Gogolla, N.; Leblanc, J.J.; Quast, K.B.; Südhof, T.C.; Fagiolini, M.; Hensch, T.K. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 2009, 1, 172–181.
- Ezhilarasan, D.; Mani, U. Valproic acid induced liver injury: An insight into molecular toxicological mechanism. Environ. Toxicol. Pharmacol. 2022, 95, 103967.
- Shakya, P.; Mohanty, C.; Mohanty, C. Hepatotoxicity of Valproate on Fetal Mice Liver. IJSR-Int. J. Sci. Res. Med. Sci. 2016, 5, 79–80.
- Akta, A.; Nergız, Y.; Akku, M.; Nasır, Y. The effects of valproic acid on renal corpuscle of pregnant rats and protective role of folic acid and vitamin E. Afr. J. Biotechnol. 2010, 9, 5605–5610.
- Raza, M.; Al Shabanah, O.; Mohammed al-Bekairi, A.; Qureshi, S. Pathomorphological changes in mouse liver and kidney during prolonged valproate administration. Int. J. Tissue React. 2000, 22, 15–21.
- Michaelis, M.; Michaelis, U.R.; Fleming, I.; Suhan, T.; Cinatl, J.; Blaheta, R.A.; Hoffmann, K.; Kotchetkov, R.; Busse, R.; Nau, H.; et al. Valproic Acid Inhibits Angiogenesis in Vitro and in Vivo. Mol. Pharmacol. 2004, 65, 520–527.
- Nau, H.; Hauck, R.S.S.; Ehlers, K. Valproic Acid-Induced Neural Tube Defects in Mouse and Human: Aspects of Chirality, Alternative Drug Development, Pharmacokinetics and Possible Mechanisms. Pharmacol. Toxicol. 1991, 69, 310–321.
- Avagliano, L.; Massa, V.; George, T.M.; Qureshy, S.; Bulfamante, G.P.; Finnell, R.H. Overview on Neural tube defects: From development to physical characteristics. Birth Defects Res. 2019, 111, 1455.
- Genton, P.; Semah, F.; Trinka, E. Valproic acid in epilepsy: Pregnancy-related issues. Drug Saf. 2006, 29, 1–21.
- Hansen, J.M.; Lucas, S.M.; Ramos, C.D.; Green, E.J.; Nuttall, D.J.; Clark, D.S.; Marchant, E.D.; Hancock, C.R.; Piorczynski, T.B. Valproic acid promotes SOD2 acetylation: A potential mechanism of valproic acid-induced oxidative stress in developing systems Valproic acid promotes SOD2 acetylation: A potential mechanism of valproic acid-induced oxidative stress in developing systems Valproic acid promotes SOD2 acetylation: A potential mechanism of valproic acid-induced oxidative stress in developing systems. Free Radic. Res. 2021, 55, 1130–1144.
- Lundberg, Y.W.; Cabrera, R.M.; Greer, K.A.; Zhao, J.; Garg, R.; Finnell, R.H. Mapping a chromosomal locus for valproic acid-induced exencephaly in mice. Mamm. Genome 2004, 15, 361–369.
- Menegola, E.; Di Renzo, F.; Broccia, M.L.; Prudenziati, M.; Minucci, S.; Massa, V.; Giavini, E. Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2005, 74, 392–398.
- Wegner, C.; Nau, H. Diurnal variation of folate concentrations in mouse embryo and plasma: The protective effect of folinic acid on valproic-acid-induced teratogenicity is time dependent. Reprod. Toxicol. 1991, 5, 465–471.
- Ehlers, K.; Elmazar, M.M.A.; Hau, A.H. Methionine Reduces the Valproic Acid-Induced Spina Bifida Rate in Mice without Altering Valproic Acid Kinetics. J. Nutr. 1996, 126, 67–75.
- Błaszczyk, B.; Miziak, B.; Pluta, R.; Czuczwar, S.J. Epilepsy in Pregnancy-Management Principles and Focus on Valproate. Int. J. Mol. Sci. 2022, 23, 1369.
- Chaliha, D.; Albrecht, M.; Vaccarezza, M.; Takechi, R.; Lam, V.; Al-Salami, H.; Mamo, J. A Systematic Review of the Valproic-Acid-Induced Rodent Model of Autism. Dev. Neurosci. 2020, 42, 12–48.
- Wagner, G.C.; Reuhl, K.R.; Cheh, M.; McRae, P.; Halladay, A.K. A new neurobehavioral model of autism in mice: Pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord. 2006, 36, 779–793.
- Arndt, T.L.; Stodgell, C.J.; Rodier, P.M. The teratology of autism. Int. J. Dev. Neurosci. 2005, 23, 189–199.
- Fereshetyan, K.; Chavushyan, V.; Danielyan, M.; Yenkoyan, K. Assessment of behavioral, morphological and electrophysiological changes in prenatal and postnatal valproate induced rat models of autism spectrum disorder. Sci. Rep. 2021, 11, 23471.
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Nelson, S.; Romano, J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996, 370, 247–261.
- Ingram, J.L.; Peckham, S.M.; Tisdale, B.; Rodier, P.M. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 2000, 22, 319–324.
- Bauman, M. Neuroanatomic observation of the brain in autism. In The Neurobiology of Autism; Johns Hopkins UP: Baltimore, MD, USA, 1994; pp. 119–145.
- Bailey, A.; Phillips, W.; Rutter, M. Autism: Towards an Integration of Clinical, Genetic, Neuropsychological, and Neurobiological Perspectives. J. Child Psychol. Psychiatry 1996, 37, 89–126.
- Weinstein-Fudim, L.; Ergaz, Z.; Turgeman, G.; Yanai, J.; Szyf, M.; Ornoy, A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? Int. J. Mol. Sci. 2019, 20, 5278.
- Ornoy, A.; Weinstein-Fudim, L.; Becker, M.; Gorobets, D.; Szyf, M. Gender specific neurobehavioral and gene expression changes in a valproic acid (VPA)–induced mouse model of autistic like behavior and correction by S-adenosylmethionine (SAMe). In Sex, Gender, and Epigenetics: From Molecule to Bedside; Legato, M.J., Feldberg, D., Glezerman, M., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 180–197. ISBN 9780128239384.
- Sawada, K.; Kamiya, S.; Aoki, I. Neonatal valproic acid exposure produces altered gyrification related to increased parvalbumin-immunopositive neuron density with thickened sulcal floors. PLoS ONE 2021, 16, e0250262.
- Ornoy, A.; Weinstein-Fudim, L.; Becker, M. SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies. Pharmaceuticals 2022, 15, 192.
- Wang, J.; Barstein, J.; Ethridge, L.E.; Mosconi, M.W.; Takarae, Y.; Sweeney, J.A. Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord. 2013, 5, 24.
- Welsh, J.P.; Ahn, E.S.; Placantonakis, D.G. Is autism due to brain desynchronization? Int. J. Dev. Neurosci. 2005, 23, 253–263.
- Lenart, J.; Augustyniak, J.; Lazarewicz, J.W.; Zieminska, E. Altered expression of glutamatergic and GABAergic genes in the valproic acid-induced rat model of autism: A screening test. Toxicology 2020, 440, 152500.
- Feleke, R.; Jazayeri, D.; Abouzeid, M.; Powell, K.L.; Srivastava, P.K.; O’Brien, T.J.; Jones, N.C.; Johnson, M.R. Integrative genomics reveals pathogenic mediator of valproate-induced neurodevelopmental disability. Brain 2022, 145, 3832–3842.
- Zhang, R.; Zhou, J.; Ren, J.; Sun, S.; Di, Y.; Wang, H.; An, X.; Zhang, K.; Zhang, J.; Qian, Z.; et al. Transcriptional and splicing dysregulation in the prefrontal cortex in valproic acid rat model of autism. Reprod. Toxicol. 2018, 77, 53–61.
- Huang, J.Y.; Tian, Y.; Wang, H.J.; Shen, H.; Wang, H.; Long, S.; Liao, M.H.; Liu, Z.R.; Wang, Z.M.; Li, D.; et al. Functional Genomic Analyses Identify Pathways Dysregulated in Animal Model of Autism. CNS Neurosci. Ther. 2016, 22, 845–853.
- Guerra, M.; Medici, V.; Weatheritt, R.; Corvino, V.; Palacios, D.; Geloso, M.C.; Farini, D.; Sette, C. Fetal exposure to valproic acid dysregulates the expression of autism-linked genes in the developing cerebellum. Transl. Psychiatry 2023, 13, 114.
- Wegner, C.; Nau, H. Alteration of embryonic folate metabolism by valproic acid during organogenesis: Implications for mechanism of teratogenesis. Neurology 1992, 42, 17–24.
- Hansen, D.K.; Grafton, T.F.; Dial, S.L.; Gehring, T.A.; Siitonen, P.H. Effect of supplemental folic acid on valproic acid-induced embryotoxicity and tissue zinc levels in vivo. Teratology 1995, 52, 277–285.
- Semmler, A.; Frisch, C.; Bleul, C.; Smith, D.; Bigler, L.; Prost, J.C.; Blom, H.; Linnebank, M. Intrauterine valproate exposure is associated with alterations in hippocampal cell numbers and folate metabolism in a rat model of valproate teratogenicity. Seizure 2017, 46, 7–12.
- Semmler, A.; Frisch, C.; Smith, D.; Blom, H.; Linnebank, M. The ratio of S-adenosylmethione and S-adenosyl-homocysteine is increased in the brains of newborn rats in a model of valproic acid teratogenicity. Toxicology 2012, 293, 132–133.
- Bokor, S.; Vass, R.A.; Funke, S.; Ertl, T.; Molnár, D. Epigenetic Effect of Maternal Methyl-Group Donor Intake on Offspring’s Health and Disease. Life 2022, 12, 609.
- Clare, C.E.; Brassington, A.H.; Kwong, W.Y.; Sinclair, K.D. One-Carbon Metabolism: Linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu. Rev. Anim. Biosci. 2019, 7, 263–287.
- Jacob, B.R.; Piorczynski, T.B.; Hansen, J.M. VPA Inhibits P19 Neural Differentiation through Redox Dysregulation and Oxidative Stress. Free Radic. Biol. Med. 2017, 112, 190.
- Lucas, S.; Hansen, J. Valproic acid inhibits superoxide dismutase activity in mouse P19 cells. Free Radic. Biol. Med. 2018, 128, S118.
- Piorczynski, T.; Larsen, M.; Hansen, J. The protective effects of Nrf2 against valproic acid in differentiating P19 cells. Free Radic. Biol. Med. 2018, 128, S118–S119.
- Piorczynski, T.B.; Larsen, M.W.; Lee, S.J.; Hansen, J.M. NRF2 activation protects against valproic acid-induced disruption of neurogenesis in P19 cells. Differentiation 2022, 123, 18–29.
- Furugen, A.; Kanno, Y.; Ohyama, N.; Kurosawa, Y.; Jinno, N.; Narumi, K.; Iseki, K.; Kobayashi, M. Effects of valproate, an HDAC inhibitor, on the expression of folate carriers and folate metabolism-related genes in the placenta of rats. Drug Metab. Pharmacokinet. 2021, 40, 100409.
- Reynolds, E.H. Antiepileptic drugs and folate revisited. Epilepsy Behav. 2020, 112, 107336.
- Reynolds, E.H.; Green, R. Valproate and folate: Congenital and developmental risks. Epilepsy Behav. 2020, 108, 107068.
- Copp, A.J.; Stanier, P.; Greene, N.D.E. Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurol. 2013, 12, 799–810.
- Heseker, H.B.; Mason, J.B.; Selhub, J.; Rosenberg, I.H.; Jacques, P.F. Not all cases of neural-tube defect can be prevented by increasing the intake of folic acid. Br. J. Nutr. 2008, 102, 173–180.
- Lawrence, M.A.; Chai, W.; Kara, R.; Rosenberg, I.H.; Scott, J.; Tedstone, A. Examination of selected national policies towards mandatory folic acid fortification. Nutr. Rev. 2009, 67, S73–S78.
- Petersen, J.M.; Smith-Webb, R.S.; Shaw, G.M.; Carmichael, S.L.; Desrosiers, T.A.; Nestoridi, E.; Darling, A.M.; Parker, S.E.; Politis, M.D.; Yazdy, M.M.; et al. Periconceptional intakes of methyl donors and other micronutrients involved in one-carbon metabolism may further reduce the risk of neural tube defects in offspring: A United States population-based case-control study of women meeting the folic acid recommendations. Am. J. Clin. Nutr. 2023, 118, 720–728.
- Shaw, G.M.; Carmichael, S.L.; Yang, W.; Selvin, S.; Schaffer, D.M. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am. J. Epidemiol. 2004, 160, 102–109.
- Williams, L.J.; Mai, C.T.; Edmonds, L.D.; Shaw, G.M.; Kirby, R.S.; Hobbs, C.A.; Sever, L.E.; Miller, L.A.; John Meaney, F.; Levitt, M. Prevalence of spina bifida and anencephaly during the transition to mandatory folic acid fortification in the United States. Teratology 2002, 66, 33–39.
- Lin, D.W.; Chung, B.P.; Kaiser, P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J. Cell Sci. 2014, 127, 50–59.
- Akimova, D.; Wlodarczyk, B.J.; Lin, Y.; Ross, M.E.; Finnell, R.H.; Chen, Q.; Gross, S.S. Metabolite profiling of whole murine embryos reveals metabolic perturbations associated with maternal valproate-induced neural tube closure defects. Birth Defects Res. 2017, 109, 106–119.
- D’Souza, S.W.; Glazier, J.D. Homocysteine Metabolism in Pregnancy and Developmental Impacts. Front. Cell Dev. Biol. 2022, 10, 802285.
- Zhu, S.; Ni, G.; Sui, L.; Zhao, Y.; Zhang, X.; Dai, Q.; Chen, A.; Lin, W.; Li, Y.; Huang, M.; et al. Genetic Polymorphisms in Enzymes Involved in One-Carbon Metabolism and Anti-epileptic Drug Monotherapy on Homocysteine Metabolism in Patients with Epilepsy. Front. Neurol. 2021, 12, 683275.
- Zhao, W.; Mosley, B.S.; Cleves, M.A.; Melnyk, S.; James, S.J.; Hobbs, C.A. Neural tube defects and maternal biomarkers of folate, homocysteine, and glutathione metabolism. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 230–236.
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901.
- Wu, G.; Nan, C.; Rollo, J.C.; Huang, X.; Tian, J. Sodium valproate-induced congenital cardiac abnormalities in mice are associated with the inhibition of histone deacetylase. J. Biomed. Sci. 2010, 17, 16.
- Leung, C.S.; Rosenzweig, S.J.; Yoon, B.; Marinelli, N.A.; Hollingsworth, E.W.; Maguire, A.M.; Cowen, M.H.; Schmidt, M.; Imitola, J.; Gamsiz Uzun, E.D.; et al. Dysregulation of the chromatin environment leads to differential alternative splicing as a mechanism of disease in a human model of autism spectrum disorder. Hum. Mol. Genet. 2023, 32, 1634–1646.
- Danielsson, B.R.; Danielsson, C.; Nilsson, M.F. Embryonic cardiac arrhythmia and generation of reactive oxygen species: Common teratogenic mechanism for IKr blocking drugs. Reprod. Toxicol. 2007, 24, 42–56.
- Echefu, B.E.; Bello, A.; Musa, S.A.; Umana, U.E. Selenium Mitigates Prenatal Lead-Induced Toxicity on Cerebral Cortex of Wistar Rats Pups. Res. Sq. 2022. preprint.
- Echefu, B.; Musa, S.; Umana, U.; Haman, W.; Abel, P. Selenium impact assessment on brains of prenatally lead exposed Wistar rats. IBRO Rep. 2019, 6, S338.
- Verrotti, A.; Scardapane, A.; Franzoni, E.; Manco, R.; Chiarelli, F. Increased oxidative stress in epileptic children treated with valproic acid. Epilepsy Res. 2008, 78, 171–177.
- Ornoy, A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod. Toxicol. 2007, 24, 31–41.
- Finsterer, J. Toxicity of antiepileptic drugs to mitochondria. Handb. Exp. Pharmacol. 2017, 240, 473–488.
- Komulainen, T.; Lodge, T.; Hinttala, R.; Bolszak, M.; Pietilä, M.; Koivunen, P.; Hakkola, J.; Poulton, J.; Morten, K.J.; Uusimaa, J. Sodium valproate induces mitochondrial respiration dysfunction in HepG2 in vitro cell model. Toxicology 2015, 331, 47–56.
- Da Costa, R.M.; Karmirian, K.; Rehen, S.K.; Kastrup Rehen, S. Deformation of Mitochondrial Cristae in Human Neural Progenitor Cells Exposed to Valproic Acid. Acad. Bras. Cienc. 2018, 90, 2223–2232.
- Sendrowski, K.; Sobaniec, W.; Sobaniec, P.; Sobaniec-Lotowska, M.E. Ultrastructural study of hippocampal cortex neurons in an experimental model of valproate encephalopathy. Folia Histochem. Cytobiol. 2013, 51, 31–37.
- Hroudova, J.; Fisar, Z. Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuro Endocrinol. Lett. 2010, 31, 336–342.
- Kudin, A.P.; Mawasi, H.; Eisenkraft, A.; Elger, C.E.; Bialer, M.; Kunz, W.S. Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1912.
- Silva, M.F.B.; Aires, C.C.P.; Luis, P.B.M.; Ruiter, J.P.N.; Ijlst, L.; Duran, M.; Wanders, R.J.A.; Tavares de Almeida, I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. Dis. 2008, 31, 205–216.
- Cauvin, C.; Echard, A. Phosphoinositides: Lipids with informative heads and mastermind functions in cell division. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 832–843.
- Greene, N.D.E.; Leung, K.Y.; Copp, A.J. Inositol, neural tube closure and the prevention of neural tube defects. Birth Defects Res. 2017, 109, 68–80.
- Posor, Y.; Eichhorn-Grünig, M.; Haucke, V. Phosphoinositides in endocytosis. Biochim. Biophys. Acta 2015, 1851, 794–804.
- Tsujita, K.; Itoh, T. Phosphoinositides in the regulation of actin cortex and cell migration. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 824–831.
- Yanai, J.; Vigoda, M.J.; Ornoy, A. Reversal of neurobehavioral teratogenicity in animal models and human: Three decades of progress. Brain Res. Bull. 2019, 150, 328–342.
- De Jong, E.; Doedée, A.M.C.M.; Reis-Fernandes, M.A.; Nau, H.; Piersma, A.H. Potency ranking of valproic acid analogues as to inhibition of cardiac differentiation of embryonic stem cells in comparison to their in vivo embryotoxicity. Reprod. Toxicol. 2011, 31, 375–382.
- Blaheta, R.; Nau, H.; Michaelis, M.; Cinatl, J., Jr. Valproate and valproate-analogues: Potent tools to fight against cancer. Curr. Med. Chem. 2002, 9, 1417–1433.
- Eickholt, B.J.; Towers, G.J.; Ryves, W.J.; Eikel, D.; Adley, K.; Ylinen, L.M.J.; Chadborn, N.H.; Harwood, A.J.; Nau, H.; Williams, R.S.B. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, glycogen synthase kinase-3β inhibition, and viral replication: A screening approach for new bipolar disorder drugs derived from the valproic acid core structure. Mol. Pharmacol. 2005, 67, 1426–1433.
- Heinz, N. VPA_terato_study. Fundam. Appl. Toxicol. 1986, 6, 662–668.
- Mishra, M.K.; Kukal, S.; Paul, P.R.; Bora, S.; Singh, A.; Kukreti, S.; Saso, L.; Muthusamy, K.; Hasija, Y.; Kukreti, R. Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules 2022, 27, 104.
- Tang, W.; Abbott, F.S. Bioactivation of a Toxic Metabolite of Valproic Acid, (E)-2-Propyl-2,4-pentadienoic Acid, via Glucuronidation. LC/MS/MS Characterization of the GSH-Glucuronide Diconjugates. Chem. Res. Toxicol. 1996, 9, 517–526.
- Fathe, K.; Palacios, A.; Finnell, R.H. Brief report novel mechanism for valproate-induced teratogenicity. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 592–597.
- Nakhal, M.M.; Jayaprakash, P.; Aburuz, S.; Sadek, B.; Akour, A. Canagliflozin Ameliorates Oxidative Stress and Autistic-like Features in Valproic-Acid-Induced Autism in Rats: Comparison with Aripiprazole Action. Pharmaceuticals 2023, 16, 769.
- Zhang, Y.H.; Wang, T.; Li, Y.F.; Deng, Y.N.; He, X.L.; Wang, L.J. N-acetylcysteine improves autism-like behavior by recovering autophagic deficiency and decreasing Notch-1/Hes-1 pathway activity. Exp. Biol. Med. 2023, 248, 966–978.
- Yamaguchi, H.; Hara, Y.; Ago, Y.; Takano, E.; Hasebe, S.; Nakazawa, T.; Hashimoto, H.; Matsuda, T.; Takuma, K. Environmental enrichment attenuates behavioral abnormalities in valproic acid-exposed autism model mice. Behav. Brain Res. 2017, 333, 67–73.
- Kumar, H.; Sharma, B. Minocycline ameliorates prenatal valproic acid induced autistic behaviour, biochemistry and blood brain barrier impairments in rats. Brain Res. 2016, 1630, 83–97.
- Luo, L.; Chen, J.; Wu, Q.; Yuan, B.; Hu, C.; Yang, T.; Wei, H.; Li, T. Prenatally VPA exposure is likely to cause autistic-like behavior in the rats offspring via TREM2 down-regulation to affect the microglial activation and synapse alterations. Environ. Toxicol. Pharmacol. 2023, 99, 104090.


