Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bernhard Biersack | -- | 14927 | 2024-03-13 17:56:17 | | | |
| 2 | Catherine Yang | Meta information modification | 14927 | 2024-03-14 01:54:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Biersack, B.; Tahtamouni, L.; Höpfner, M. Receptor Tyrosine Kinases in BRAF Mutant Cancers. Encyclopedia. Available online: https://encyclopedia.pub/entry/56229 (accessed on 26 July 2026).
Biersack B, Tahtamouni L, Höpfner M. Receptor Tyrosine Kinases in BRAF Mutant Cancers. Encyclopedia. Available at: https://encyclopedia.pub/entry/56229. Accessed July 26, 2026.
Biersack, Bernhard, Lubna Tahtamouni, Michael Höpfner. "Receptor Tyrosine Kinases in BRAF Mutant Cancers" Encyclopedia, https://encyclopedia.pub/entry/56229 (accessed July 26, 2026).
Biersack, B., Tahtamouni, L., & Höpfner, M. (2024, March 13). Receptor Tyrosine Kinases in BRAF Mutant Cancers. In Encyclopedia. https://encyclopedia.pub/entry/56229
Biersack, Bernhard, et al. "Receptor Tyrosine Kinases in BRAF Mutant Cancers." Encyclopedia. Web. 13 March, 2024.
Copy Citation
BRAF (B-Raf, B-rapidly accelerated fibrosarcoma) mutations are clinically relevant in melanoma, non-small-cell lung cancer (NSCLC), colorectal carcinoma (CRC), and other cancers. Patients suffering from BRAF mutant cancers are experiencing a considerably poor prognosis. Receptor tyrosine kinases (RTKs), which are prominent anticancer drug targets in their own right, play a crucial role in the development of drug resistance to BRAF inhibitors and the reactivation of MAPK/ERK signal transduction, as well as the establishment of bypassing signaling pathways.
receptor tyrosine kinase
growth factor receptors
BRAF mutant cancer
1. EGFR, HER2, and HER3 (ErbB Receptors)
1.1. ErbB Receptors, Kinase Inhibitors, Main Mechanisms, and Targets
EGFR (ErbB1) is a transmembrane receptor of the ErbB subfamily for epidermal growth factors (EGFs and other EGF family growth factors such as neuregulins/NRGs), and activating mutations of EGFR play a crucial role in the development and progression of various cancers such as lung cancer, colorectal carcinoma, head-and-neck cancers, and glioblastoma [1][2]. Upon binding of EGF to the extracellular EGFR binding sites, the EGFR proteins form active dimers (homodimers or heterodimers with other ErbB proteins), which are able to transmit the EGF signal via docking and adapter proteins (e.g., Grb2 and SOS) to prenylated membrane-bound Ras proteins [3]. PI3K/AKT signaling is another oncogenic signaling pathway activated by EGFR [4]. HER2 (human epidermal growth factor 2, ErbB2), HER3 (ErbB3), and HER4 (ErbB4) are other members of the ErbB family of RTKs with particular significance in breast cancer, which form heterodimers with other ErbB proteins (e.g., HER2/HER3 receptors), including EGFR [5].
The development of the first-generation EGFR inhibitors, gefitinib and erlotinib, was a landmark in targeted tumor therapy [6]. Irreversible second-generation inhibitors such as afatinib and third-generation inhibitors such as osimertinib, as well as the EGFR-targeting antibody cetuximab, complement the current arsenal of clinically applied EGFR inhibitors [7]. Lapatinib is a prominent dual EGFR and HER2 inhibitor, while afatinib and neratinib inhibit EGFR, HER2, and HER4 [5]. However, resistance mechanisms such as activating EGFR mutations (e.g., the T790M mutation) and upregulating bypass signaling pathways pose a considerable clinical problem [8]. Such adaptive responses to TKIs leading to kinome reprogramming need to be thoroughly understood to overcome cancer drug resistance [9]. For instance, the formation of drug-tolerant persister cells upon treatment with BRAF and MEK inhibitors was influenced by RTK activation kinetics, and sustained RTK activation led to higher expression of ERK target genes despite BRAFi (BRAF inhibitor) and MEKi treatment [10].
EGFR was soon identified as a crucial factor that conveys resistance to BRAF inhibitor treatment in BRAF mutant cancers. Inhibition of BRAFV600E by vemurafenib interferes with the ERK-controlled negative feedback loop of MAPK signaling, which upregulates EGFR, followed by MAPK reactivation and vemurafenib resistance [11]. Gefitinib, afatinib, and lapatinib in combination with AKT inhibitors and vemurafenib (triple combination) were found to be active against BRAF mutant melanomas [12]. In BRAF mutant melanoma cells, the combination of gefitinib and vemurafenib led to additive effects accompanied by the suppression of colony formation and migration in preclinical studies. In contrast, wild-type BRAF melanoma cells showed considerable resistance to gefitinib treatment [13]. The reactivation of MAPK signaling in BRAF mutant CRCs treated with vemurafenib also occurred via EGFR, as evident by the activation of Ras and CRAF. In addition, AKT activation was observed upon vemurafenib treatment. Yet, the combination of the EGFR and BRAF inhibitors erlotinib and vemurafenib efficiently suppressed (BRAF mutant) HT-29 and WiDr CRC growth in vitro and in vivo [11]. Moreover, the triple combination of BRAF, MEK, and EGFR inhibitors exhibited promising results in various BRAF mutant CRC cell lines with acquired BRAF inhibitor resistance, which displayed gene amplification and/or mutation of the EGFR and KRAS genes [14]. Recently, CRISPR-Cas experiments underlined the potential of triple targeting of BRAF, MEK, and EGFR for a more efficient therapy of BRAF mutant CRC [15]. In addition, BRAF mutant melanoma cells with acquired vemurafenib resistance and high EGFR expression levels showing increased cell migration can be less susceptible to erlotinib treatment and were characterized by high expression of the immune checkpoint protein PD-L1, indicating another suitable target to cope with vemurafenib resistance in melanoma when EGFR inhibition fails [16].
The outcomes of clinical studies using the combination of BRAF/MEK inhibitors with checkpoint inhibitors were recently summarized. However, only the IMspire150 phase 3 study with advanced BRAFV600E mutant melanoma using atezolizumab (a PD-L1 inhibitor) plus vemurafenib and cobimetinib (a MEK1 inhibitor) showed promising results in terms of prolonged progression-free survival and increased 5-year survival [17]. Intrinsic resistance to BRAF inhibitor treatment in melanoma characterized by high EGFR and low HER3 levels was accompanied by a limited response to EGFR inhibitors, and targeting of the PI3K/AKT signaling pathway appeared to be more promising [18]. Yet, a case report of a BRAF mutant melanoma patient with brain metastases suggests that resistance formation by increased EGFR levels upon first-line BRAF/MEK inhibitor treatment with dabrafinib plus trametinib is reversible, and the pre-treated tumors can show clinical responses to rechallenge treatment with encorafenib plus binimetinib in the following [19].
In vitro 3D BRAF mutant CRC models treated with vemurafenib and gefitinib revealed an important role of HGFR (hepatocyte growth factor receptor, also known as MET) activation in the establishment of BRAF and EGFR inhibitor resistance via AKT signaling activation, which was less pronounced in 2D tumor models [20]. In addition to PI3K/AKT signaling, other pathways such as JAK/STAT can be involved in EGFR-mediated BRAF inhibitor resistance in BRAF mutant melanoma, while synergistic effects of EGFR and HER2 inhibitor lapatinib in combination with the BRAF inhibitor PLX4720 can occur independently from AKT signaling and MAPK reactivation. Here, the synergistic effects of the dual EGFR/HER2 inhibitor lapatinib were similar to PLX4720 combinations with masatinib but superior to the effects of gefitinib, a selective EGFR inhibitor without activity against HER2, indicating a beneficial role of HER family kinase inhibition [21]. Lapatinib was likewise efficient in eradicating BRAF mutant CRC cells in combination with the AKT inhibitor MK2206, which revealed distinct synergistic effects [22]. Moreover, HER inhibition by lapatinib blocked the MAPK rebound effect in papillary thyroid carcinoma and sensitized BRAFV600E thyroid cancer cells to BRAF/MEK inhibitor treatment. Further tests showed that lapatinib also augmented radioiodine uptake, which is significant since BRAFV600E mutant cells are resistant to radioiodine therapy [23]. The EGFR/HER2 inhibitor afatinib was especially active against COLO-205 CRC cells, which display high HER2 expression, and the combination of afatinib with vemurafenib showed additive effects on BRAFV600E CRC [24]. The irreversible EGFR/HER2/HER4 and MAP4K (Ste20 family serine–threonine kinase) inhibitor neratinib eliminated BRAFV600E cutaneous melanoma cells, showed synergistic effects with HDAC inhibitors, and led to ROS-dependent autophagosome formation [25]. Pan-ErbB inhibition by canertinib led to apoptosis in BRAF mutant melanoma cells and blocked EGF- and NRG1-induced ErbB signaling. Canertinib blocked EGF-induced AKT and inhibited STAT3 phosphorylation in the absence of EGF [26]. Resistant BRAF mutant canine transitional cell carcinoma of the bladder was re-sensitized to vemurafenib treatment in combination with the pan-ErbB inhibitor sapitinib [27]. The relevance of EGFR was also shown in studies with next-generation BRAF inhibitors. PLX8394 is a BRAF dimer inhibitor, which can evade MAPK reactivation by vemurafenib treatment, and was active both against BRAFV600E and non-V600 lung adenocarcinomas as well as against cells with truncated vemurafenib-insensitive BRAFV600E. Acquired PLX8394 resistance in lung adenocarcinoma was mediated by EGFR-RAS-mTOR signaling and was overcome by a combination of PLX8394 with an EGFR inhibitor (erlotinib) or an mTOR inhibitor (everolimus) [28]. Based on the central role of EGFR in Ras/MAPK signaling and BRAF inhibitor resistance formation, the first-in-class dual RAF/EGFR inhibitor lifirafenib (BGB-283) was developed, which reversibly inhibits RAF dimers of wild-type ARAF, BRAF, and CRAF, as well as BRAFV600E and EGFR, which was accompanied by distinctly increased activity against BRAF mutant CRC when compared with vemurafenib and dabrafenib [29]. Lifirafenib was effective against vemurafenib-insensitive non-V600 BRAF mutant lung cancers, whose MAPK re-activation mechanisms extremely rely on EGFR [30]. Lifirafenib showed promising results in a phase 1 study with patients suffering from solid tumors, and responses were observed in BRAF mutant melanoma, thyroid cancer, and ovarian cancer [31]. Lifirafenib successfully suppressed MAPK reactivation and in vivo KRAS mutant tumor growth in combination with the MEK inhibitor selumetinib, and a clinical phase 1b/2 study of lifirafenib in combination with the MEK inhibitor mirdametinib was launched for solid tumors with MAPK aberrations, including KRAS and RAF mutations [32].
While EGFR activation poses a resistance mechanism for BRAF inhibitors, mutant BRAF associated with drug resistance can also appear in turn as a consequence of EGFR inhibitor therapy. Notably, the emergence of NSCLC tumors with acquired V600 and non-V600 BRAF mutations was reported, featuring pivotal resistance factors for the treatment with osimertinib, which warrants the combined application of EGFR inhibitors with BRAF and MEK inhibitors for these cases [33]. In an EGFR mutant/BRAF mutant lung adenocarcinoma patient suffering from bone metastases who developed BRAFV600E as a consequence of second-line osimertinib treatment, an osimertinib-based triple combination therapy with dabrafenib and trametinib showed impressive outcomes such as overall tumor response and complete bone pain reduction with an enduring asymptomatic state after continuation of this triple combination therapy [34]. Another NSCLC case of acquired resistance based on the BRAFV600E mutation after treatment with osimertinib exhibited tumor regression and reduced symptoms [35]. In addition, a patient suffering from EGFR (del19) mutant/BRAFV600E NSCLC with life-threatening leptomeningeal brain metastasis also responded well to this triple therapy [36]. The EGFR del19 mutation is the most frequently detected activating mutation in NSCLC that sensitizes cancer cells to EGFR TKI therapy [37]. However, the emergence of BRAF fusion genes such as BTN2A1-BRAF leading to BRAF overexpression via promoter deregulation as an acquired resistance mechanism to osimertinib treatment poses a significant problem for patients with advanced NSCLC [38].
In addition to the small-molecule EGFR inhibitors mentioned above, the anti-EGFR monoclonal antibody (mAb) cetuximab was approved for the therapy of EGFR-related cancers [39]. Thus, the effects of cetuximab on BRAF inhibitor resistance are of great importance. Treatment of BRAFV600E mutant HT-29 cells with the mTOR inhibitor PP242 increased EGFR phosphorylation/activation, and the combination of cetuximab and PP242 inhibited the growth of HT-29 xenografts more efficiently than the single drugs [40]. Although quite inactive against HT-29 CRC cells as a single drug, cetuximab augmented the cell-killing activity of peripheral blood natural killer (NK) cells against HT-29 cells [41]. In combination with dabrafenib, cetuximab induced PTEN and suppressed Src and c-Myc in BRAFV600E mutant CRC cells [42]. Non-V600 BRAF mutant lung and colorectal cancers (i.e., class III BRAF mutants) with low BRAF kinase activity were found to be especially sensitive to cetuximab, which led to the launch of clinical trials with EGFR inhibitors in non-V600 cancers (see below) [43]. Trastuzumab emtansine is an antibody drug conjugate of the HER2 targeting antibody trastuzumab with the maytansinoid tubulin inhibitor emtansine (mertansine). This conjugate was more active than cetuximab and trastuzumab against HER2-positive BRAF mutant CRC cells. The combination of trastuzumab emtansine with metformin, an antidiabetic drug that upregulates endocytic calveolin-1 expression, led to increased anticancer activity against HER2-positive BRAF mutant CRC cells and xenograft models [44]. The formation of radioiodine-refractory (RAIR) thyroid cancer is often correlated with the BRAFV600E mutation and MAPK activation. Results of a pilot clinical study with BRAF mutant RAIR thyroid cancer patients showed that the combination of vemurafenib with the anti-ErbB3 monoclonal antibody CDX-3379 increased iodine uptake by most patients and had a partial response in two patients. SWI/SNF gene mutations (e.g., in ARID2) were described as resistance factors for this therapy [45].
1.2. Additional Mechanisms and Factors of EGFR-Mediated BRAF Inhibitor Resistance
The upregulation of EGFR in BRAF inhibitor-resistant cutaneous melanoma was correlated with distinct epigenetic processes, so-called “back-seat drivers”, and a demethylation of EGFR regulatory DNA elements such as enhancers and the EGFR gene itself was observed. The epigenetic upregulation of EGFR led to the activation of PI3K/AKT signaling. However, erlotinib, in combination with a BRAF inhibitor, was able to overcome acquired BRAF inhibitor resistance based on high EGFR levels [46]. This epigenetic mode of EGFR activation comes along with the synergistic effect of neratinib in combination with HDAC inhibitors [25]. Several RTKs, including EGFR, were upregulated in BRAF mutant thyroid cancers dependent on phosphatase SHP2 (protein tyrosine phosphatase 2, a downstream factor of RTK signaling). Inhibition of SHP2 either by knockdown or by the SHP2 inhibitor SHP099 reversed late resistance to vemurafenib in BRAF mutant thyroid cancer cells by decelerating MAPK/ERK reactivation (Figure 1a) [47]. The phosphatase PTEN (phosphatase and tensin homolog) hydrolyzes phosphatidyl-inositol triphosphate PIP3, the activator of PI3K, to PIP2, thereby suppressing PI3K/AKT signaling. Combined inhibition of BRAF and EGFR upregulated PTEN and suppressed Src and c-Myc functions in BRAFV600E CRC cells [42]. The ubiquitin-like protein NEDD8 and its associated pathway can contribute to concomitant resistance to EGFR and BRAF inhibitor treatment, and suppression of NEDD8 by the NEDD8 pathway inhibitor pevonedistat combined with the EGFR inhibitors cetuximab or lapatinib efficiently blocked feedback loops in BRAF mutant CRC (Figure 1a) [48]. The combination of pevonedistat with EGFR inhibitors was crucial for anticancer activity since pevonedistat stabilized activated phospho-EGFR by suppressing its ubiquitination, leading to upregulation of EGFR signaling, which was abrogated by EGFR inhibitors. This comes along with increasing evidence of UPR (unfolded protein response) and associated Hsp70 as promising targets for the treatment of BRAFV600E mutant CRC with a poor prognosis [49].
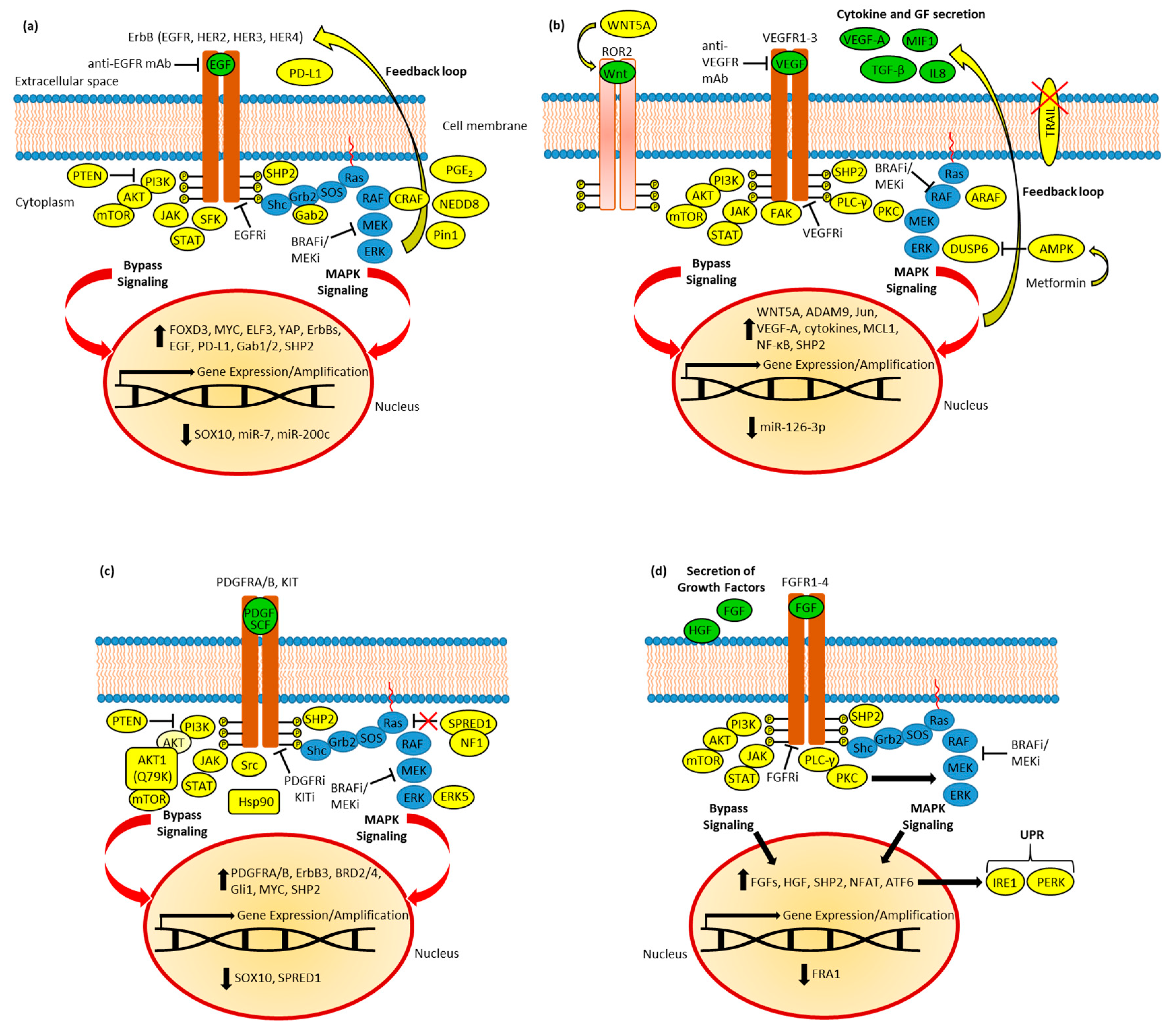
Figure 1. (a) EGFR in BRAF inhibitor (BRAFi) resistance. Resistant cells established feedback loops and bypassed signaling via AKT, JAK/STAT, and SFK signaling, accompanied by deregulation of transcription factors (SOX10, FOXD3, MYC, ELF3, and YAP) and miRNAs. Upregulated CRAF and PD-L1 were identified. Moreover, active SHP2, Pin1, and NEDD8 as well as upregulated PGE2 synthesis promote resistance. (b) VEGFR in BRAFi resistance. The feedback loop leads to increased secretion of cytokines and growth factors (GF). ARAF and SHP2, as well as metformin-induced AMPK activation (followed by DUSP6 suppression), contribute to MAPK reactivation. TRAIL apoptosis signaling is blocked in resistant cells. Bypass signaling includes AKT, STAT, and FAK; VEGF-A and cytokine production; and Wnt-activated ROR2 RTK. Resistance was accompanied by the induction of c-Jun transcription factor and the downregulation of miR-126-3p; the latter caused the upregulation of ADAM9. (c) PDGFR and KIT in BRAFi resistance. Resistant cells established feedback loops and bypassed MAPK inhibition via AKT, JAK/STAT, and Src signaling, accompanied by deregulation of transcription factors (SOX10, MYC, and Gli1) and BRD2/4. Mutant AKT1 overcomes PTEN-based AKT inhibition, while suppressed SPRED1 and induced ERK5 lead to MAPK reactivation. RTK (PDGFR and ErbB3) expression is upregulated. Active SHP2 and Hsp90 promote resistance. (d) FGFR in BRAFi resistance and BRAF mutant tumor metastasis. Feedback leads to increased expression and secretion of growth factors. SHP2 and PKC-mediated MAPK signaling contribute to MAPK reactivation. Resistance was accompanied by suppression of the FRA1 transcription factor and induction of NFAT and ATF6, the latter of which caused upregulation of UPR by activation of IRE1 and PERK.
Melanoma patient survival was reduced by prostaglandin synthase (PTGES) expression, and acquired vemurafenib resistance in BRAF mutant melanoma was associated with increased prostaglandin E2 synthesis, which, however, was accompanied by increased sensitivity for gefitinib treatment, indicating a vital role of EGFR signaling in the inflammatory lipid metabolism of vemurafenib-resistant cells [50]. A vital EGFR–SFK (Src family kinase)–STAT3 (signal transducer and activator of transcription 3) axis was identified in BRAF inhibitor-resistant tumors of melanoma patients, which drives proliferation and lung metastasis formation by BRAF mutant melanoma cells, indicating that a combination of EGFR inhibitors with Src inhibitors can be promising to circumvent BRAF inhibitor resistance (Figure 1a) [51]. The transcription factor YAP (yes-associated protein), an effector of the Hippo pathway, was associated with resistance to RAF inhibitors [52]. The adaptive resistance of BRAFV600E mutant thyroid cancer cells to vemurafenib depended on HER2 and HER3 activation upon nuclear translocation and activation of YAP (induction of the YAP-NRG1 pathway). The EGFR/HER2 inhibitor lapatinib re-sensitized resistant cells to vemurafenib and showed promising anticancer effects in combination with the YAP inhibitor verteporfin. YAP activation also played a role in vemurafenib resistance in BRAF mutant melanoma, but not in BRAF mutant CRC (Figure 1a) [53]. The relevance of YAP for Ras signaling was corroborated by the fact that YAP1 was able to rescue KRAS-mediated transcriptional EMT (epithelial-to-mesenchymal transition) regulation and cell viability upon KRAS suppression via activating interaction with the transcription factor c-Fos [54]. In addition, upregulation of the transcription factor ELF3 (E26 transformation (ETS)-specific related transcription factor-3) in BRAF mutant thyroid cancer cells established a stable positive feedback loop with MAPK signaling by induction of HER2 and HER3 expression, which was associated with thyroid cancer progression [55]. Suppression of the SOX family transcription factor SOX10 (sex-determining region Y-box 10) induced TGF-β signaling, followed by activation of EGFR in BRAF inhibitor-resistant melanomas [56]. The transcription factor FOXD3 (forkhead box D3) upregulated HER3 as a resistance mechanism following RAF/MEK inhibitor treatment [57]. In BRAF mutant CRC cells, vemurafenib increased the levels of EGFR, HER2, and HER3, but also the expression of the Grb2-associated binders Gab1 and Gab2, two important enhancers of ErbB signal transduction downstream of RTKs. Gab2 activation was directly connected with BRAFV600E suppression, and Gab2-mediated induction of the already-described phosphatase SHP2 was important for the formation of vemurafenib resistance [58]. Suppression of the HER3-mediated positive feedback loop was achieved in BRAF mutant thyroid cancer cells upon Pin1 (peptidyl-prolyl cis/trans isomerase 1) inhibition by the Pin1 inhibitor API-1, which activated the HER3-targeting microRNA miR-20a-5p and sensitized these cells to vemurafenib (Figure 1a) [59]. Certain microRNAs were also involved in BRAF inhibitor resistance in melanomas. Suppression of EGFR-targeting miR-7 was discovered in vemurafenib-resistant cells, leading to upregulation of EGFR and CRAF and activation of MAPK and PI3K/AKT signaling [60]. In addition, cells with acquired BRAF inhibitor resistance downregulated miR-200c, accompanied by the induction of Bmi1, leading to the activation of the MAPK and PI3K/AKT pathways (Figure 1a) [61]. Generally, non-coding RNAs play pivotal roles in drug resistance to approved anticancer drugs such as platinum complexes and alkylating agents [62][63].
1.3. Clinical Trials of EGFR Inhibitors in BRAF Mutant Cancers and Adjuvant Molecules
Several clinical trials of EGFR inhibitors with cancer patients suffering from BRAF inhibitor-resistant cancers were conducted in addition to those of osimertinib and lifirafenib already mentioned above [31][32][33][34][35][36][38]. In particular, the outcome of EGFR inhibitors in BRAF mutant metastatic CRC was promising [64]. The important BEACON phase 3 study with BRAFV600E mutant metastatic CRC showed that cetuximab plus encorafenib showed acceptable safety and led to prolonged overall and progression-free survival when compared with standard therapy [65]. This was corroborated by a recent case report of a 75-year-old patient suffering from advanced pretreated BRAFV600E CRC with liver metastases, which revealed that the application of cetuximab in combination with vemurafenib and the topoisomerase I inhibitor irinotecan led to a complete response and showcased the synergy of BRAF inhibition with EGFR inhibition in the therapy of BRAFV600E CRC [66]. Recently, the combination of the two orally available drugs erlotinib and vemurafenib was studied in BRAFV600E CRC patients (phase 1b/2 EVICT study), and the combination therapy exhibited an overall response rate of 32% without dose-limiting toxicities [67]. A phase 2 study of cetuximab plus irinotecan as a “re-challenge” third-line therapy of CRC patients pretreated with second-line bevacizumab plus FOLFOX and first-line cetuximab plus FOLFIRI accompanied by Ras/BRAF-mutation was launched recently, The results of this study will be reported by 2026 [68]. The previous SWOG phase 2 study confirmed the superior effects of this triple regimen when compared with cetuximab plus irinotecan in BRAF mutant metastatic CRC; however, the addition of vemurafenib increased some grade III/IV adverse effects such as anemia, neutropenia, and nausea/vomiting [69]. A recently published Chinese study on the combination of vemurafenib, irinotecan, and cetuxamib (VIC) as a first-line therapy in unresectable BRAFV600E mutant CRC revealed prolonged overall response rate and disease control of VIC therapy when compared with chemotherapy (FOLFOX, FOLFIRI, XELOX, and FOLFOXIRI) or chemotherapy plus bevacizumab, indicating VIC as a suitable first-line therapy for this BRAF mutant CRC patient subgroup [70].
In class III BRAF mutant cancer cells, heterodimers of BRAF with CRAF induced Ras/MAPK signal transduction in a manner that was strongly EGFR- and Ras-dependent, and several clinical trials confirmed that cetuximab and panitumumab (another anti-EGFR mAb) are beneficial for the treatment of class III BRAF mutant advanced or metastatic CRCs [71][72]. The case report of a non-V600 metastatic CRC patient who experienced progressing disease upon several lines of chemotherapy (5-fluorouracil, oxaliplatin, irinotecan, and trifluridine) revealed a remarkable response upon panitumumab monotherapy, including shrinkage of metastases in the liver, adrenal gland, and retroperitoneal lymph nodes [73]. The study of BRAF mutant microsatellite stable metastatic CRC patients with primary resistance to EGFR/BRAF therapy showed an amplification of genes coding the cell cycle-regulating cyclins D1, D2, and D3, which might pave the way for the development of improved therapies for EGFR/BRAF-insensitive patient sub-groups [74]. Meta-analyses showed that combinations of BRAF and MEK inhibitors were superior to BRAF and EGFR inhibitor treatments in class II non-V600 BRAF mutant cancers but inferior in class III mutant tumors [75].
In addition to EGFR inhibitors, adjuvant compounds such as vitamins and dietary polyphenols may contribute to the treatment of BRAF inhibitor-resistant cancers via EGFR targeting. Ascorbic acid (vitamin C) suppressed MAPK and AKT signaling in both wild-type and BRAF mutant thyroid cancer cells by forming reactive oxygen species (ROS) and reduced thyroid cancer growth in vivo at high doses of 4 g/kg twice per day (i.p.). While in the BRAF mutant cancer, ascorbic acid blocked the fuel for MAPK signaling by depletion of ATP and suppressed EGF release and EGFR phosphorylation in the BRAF wild-type cells. In addition, ascorbic acid suppressed AKT through forced proteasomal degradation in wild-type and BRAF mutant cells [76]. Dietary polyphenols possess multiple mechanisms to tackle cancer drug resistance [77]. The natural polyphenol curcumin from Curcuma longa rhizomes downregulated EGFR and suppressed MAPK signaling in pancreatic and lung adenocarcinoma cells in a COX-2-dependent way [78]. In CRC cells treated with curcumin, downregulation of EGFR gene expression as well as suppression of EGFR phosphorylation were observed [79][80][81]. To investigate the effects of curcumin on BRAF mutant CRC, curcumin micelles (240 mg/kg) in drinking water were administered to an intestine-specific BRAF mutant murine model (BRAFV637E/+/Villin-CreERT2/+) for 14 months. Curcumin strongly prevented BRAF mutant CRC formation and led to reduced CRC numbers when compared with untreated mice [82]. The stilbene-based dietary polyphenol resveratrol, which naturally occurs in grapes and various berries, efficiently broke vemurafenib resistance in BRAFV600E melanoma cells associated with increased phospho-AKT levels by AKT dephosphorylation, both as a single compound and in combination with vemurafenib [83]. Resveratrol also suppressed STAT3 signaling in BRAF mutant THJ-16T (MKRN1-BRAF fusion mutation) and THJ-21T (BRAFV600E point mutation) anaplastic thyroid cancer cells via phospho-STAT3 downregulation, accompanied by suppressed MAPK signaling and reduced phosphorylation of BRAF, MAPK, and ERK. In combination with the approved BRAF/MEK-targeting drugs dabfafenib and trametinib, which induced STAT3 activation in treated cancer cells, resveratrol was able to suppress STAT3 signaling [84]. The dietary isoflavone genistein, a natural component of soybeans (Glycine max), was already identified as an EGFR inhibitor in 1987 [85]. Genistein also inhibits the proliferation driving polo-like kinase 1 (PLK1) as well as estrogen and androgen receptors (ER and AR), and a randomized phase 2 study with genistein in prostate cancer patients showed amenable tolerance of the drug and a reduction of prostate-specific antigen (PSA) levels [86][87]. In quinol-thioether-transformed rat renal epithelial (QT-RRE) cells, which lack the tumor suppressor tuberin (a suppressor of renal tumorigenesis), levels of activated BRAF, Raf-1, and ERK were high. Notably, the application of tuberin or genistein was able to downregulate BRAF and ERK in QT-RRE cells [88].
The effects and mechanisms of the described ErbB receptor inhibitors are summarized in Table 1.
Table 1. ErbB (EGFR and HER2-4) inhibitors and their activities in BRAF mutant cancers.
| ErbB Inhibitor | Mechanisms (Cancers/Cell Lines) | Clinical Studies | References |
|---|---|---|---|
| Gefitinib | Additive effects; suppression of colony formation and migration (BRAF mutant melanoma); resistance formation by MET and AKT activation (BRAF mutant CRC) | Increased activity by upregulated prostaglandin upon acquired vemurafenib resistance (BRAF mutant melanoma) | [12][13][20][50] |
| Erlotinib | Suppression of BRAF mutant CRC growth in vitro and in vivo (HT-29 and WiDr); overcomes acquired PLX8394 resistance (lung adenocarcinoma); overcomes acquired BRAF inhibitor resistance based on high EGFR levels (cutaneous melanoma) | Overall response of 32% in combination with vemurafenib (phase 1b/2 EVICT study, BRAFV600E CRC) | [11][28][46][67] |
| Afatinib | Active in combination with AKT inhibitors and vemurafenib (BRAF mutant melanoma); additive effects in combination with vemurafenib (BRAFV600E CRC) | - | [12][24] |
| Lapatinib | Synergy combined with the BRAF inhibitor PLX4720 (BRAF inhibitor-resistant BRAF mutant melanoma), and in combination with the AKT inhibitor MK2206, which revealed distinct synergistic effects (BRAF mutant CRC cells); efficient in blocking feedback loops when combined with NEDD8 inhibitor (BRAF mutant CRC); suppression of MAPK rebound effect and re-sensitization to BRAF/MEK inhibitor treatment and radioiodine therapy (BRAFV600E thyroid cancer cells); promising anticancer effects in combination with the YAP inhibitor (BRAFV600E thyroid cancer cells) | - | [21][22][23][48][53] |
| Neratinib | Synergistic effects with HDAC inhibitors; ROS-dependent autophagosome formation (BRAFV600E cutaneous melanoma cells) | - | [25] |
| Canertinib | Apoptosis induction; blocked ErbB signaling; blocking of EGF-induced AKT; inhibition of p-STAT3 in EGF’s absence (BRAF mutant melanoma cells) | - | [26] |
| Sapitinib | Re-sensitization effect (vemurafenib-resistant BRAF mutant canine transitional cell carcinoma) | - | [27] |
| Lifirafenib | Reversible inhibition of RAF dimers of wild-type ARAF, BRAF, and CRAF, and also BRAFV600E and EGFR; higher activity than vemurafenib and dabrafenib (BRAF mutant CRC); overcomes vemurafenib resistance and suppresses EGFR-mediated MAPK re-activation (non-V600 BRAF mutant lung cancers) | Clinical responses in BRAF mutant melanoma, thyroid cancer, and ovarian cancer (phase 1); ongoing clinical phase 1b/2 study in combination with the MEK inhibitor mirdametinib with KRAS and RAF mutant solid tumors | [29][30][31][32] |
| Osimertinib | Leads to acquired V600 and non-V600 BRAF mutations (NSCLC) | Clinical responses to triple combination therapy (plus dabrafenib and trametinib), including the breach of BRAFV600E-mediated osimertinib resistance (EGFR mutant/BRAF mutant lung adenocarcinoma with bone metastases and EGFR(del19) mutant/BRAFV600E NSCLC with leptomeningeal brain metastasis); problematic BRAF fusion gene (BTN2A1-BRAF) as acquired osimertinib resistance factor (advanced NSCLC) | [33][34][35][36][38] |
| Cetuximab | Combination with the mTOR inhibitor PP242 inhibited tumor growth (BRAFV600E mutant HT-29 xenografts); increased cell killing activity of peripheral blood natural killer (NK) cells (HT-29 cells); combination with dabrafenib induced PTEN and suppressed Src and c-Myc (BRAFV600E mutant CRC cells); sensitive class III BRAF mutant cancers (lung and colorectal cancers); efficient blocking of feedback loops when combined with the NEDD8 inhibitor (BRAF mutant CRC) | Prolonged overall and progression-free survival in combination with encorafenib (BEACON phase 3 with BRAFV600E mutant metastatic CRC); complete response in combination with vemurafenib and the topoisomerase I inhibitor irinotecan (case report of advanced pretreated BRAFV600E CRC with liver metastases); ongoing phase 2 study plus irinotecan as “re-challenge” third-line therapy of pretreated Ras/BRAF mutant CRC; superior effects of this cetuximab-based triple therapy (SWOG phase 2 study, BRAF mutant metastatic CRC); prolonged overall response rate and disease control in combination with vemurafenib and irinotecan (VIC) as a first-line therapy (unresectable BRAFV600E mutant CRC); activity against class III BRAF mutants (several trials, advanced or metastatic CRCs) | [40][41][42][43][48][65][66][68][69][70][71][72] |
| Panitumumab | - | Remarkable response (case report, pretreated non-V600 metastatic CRC); activity against class III BRAF mutants (several trials, advanced or metastatic CRCs) | [71][72][73] |
| Trastuzumab emtansine | Highly active; combination with metformin upregulates endocytic calveolin-1 expression and increased anticancer activity (HER2-positive BRAF mutant CRC cells and xenograft models) | - | [44] |
| Ascorbic acid | ROS formation, ATP depletion, MAPK and AKT suppression (BRAF mutant thyroid cancer cells), and suppression of thyroid cancer growth in vivo | - | [75][76] |
| Curcumin | COX-2-dependent MAPK suppression (pancreatic and lung adenocarcinoma), suppression of p-EGFR and EGFR expression (CRC), and prevention of BRAF mutant CRC formation (murine BRAFV637E/+/Villin-CreERT2/+ model) | - | [78][79][80][81][82] |
| Resveratrol | AKT dephosphorylation (BRAFV600E melanoma) and STAT3 suppression (THJ-16T MKRN1-BRAF fusion mutation and THJ-21T BRAFV600E ATC cells) | - | [83][84] |
| Genistein | Suppression of BRAF and ERK (QT-RRE cells) | - | [88] |
2. VEGFR and BRAF Mutant Cancers
2.1. VEGFRs, Inhibitors, and Mechanisms
The vascular–endothelial growth factor receptors (VEGFRs, e.g., VEGFR-1/Flt-1, VEGFR2/KDR, and VEGFR-3/Flt-4) play an important role in tumor-associated angiogenesis and tumor progression. The corresponding VEGF signal molecules and ligands (VEGF-A, B, C, D, and placental growth factor (PlGF)) interact specifically with extracellular components (immunoglobulin-like domains) of the VEGFR family [89]. Although VGFR family proteins share high structural similarities, there are differences in terms of signal transduction and functions [90]. Upon extracellular ligand binding, changes in the intracellular domain conformation lead to VEGFR dimerization and autophosphorylation at defined tyrosine sites, followed by induction of p38/MAPK, MAPK/ERK (via PKC), and PI3K/AKT/mTOR signaling pathways. Autocrine positive feedback loops were observed for VEGFRs (VEGF released by a cancer cell activates VEGFR of the same cancer cell), and activated VEGFR2 was especially associated with these processes and signaling pathways, as well as the correlated effects on cancer progression and invasion [91].
Several small-molecule multikinase inhibitors with inhibitory activity against VEGFRs (sorafenib, sunitinib, pazopanib, vandetanib, axitinib, carbozantinib, lenvatinib, regorafenib, and ponatinib) are clinically approved for the therapy of various cancer diseases [92]. It is noteworthy that, among these inhibitors, sorafenib and regorafenib are also targeting BRAF. Fruquintinib is a selective VEGFR inhibitor that was approved in China for the therapy of metastatic CRC in 2018, and its promising activity against advanced solid tumors in US patients was disclosed recently [93][94]. The anti-VEGF-A antibody bevacizumab is clinically applied for the first-line therapy of RAS and/or BRAF mutant CRC [95].
Because of their VEGFR and BRAF inhibitory activities, sorafenib and regorafenib are of immense interest for the treatment of BRAF mutant cancers. In canine transitional cell carcinoma cells with the BRAFV595E mutation (which corresponds to human BRAFV600E), sorafenib was found to be more active than vemurafenib [96]. In BRAFV600E thyroid carcinoma cells, sorafenib induced apoptosis and suppressed MAPK and AKT signaling associated with reduced phosphorylation of VEGFR1/2/3 and other RTKs such as PDGFRB. These effects were also observed in wild-type cells treated with sorafenib, which underlines the broad beneficial effect of sorafenib on thyroid cancers [97]. Yet, in BRAFV600E CRC cells, which were sensitive to vemurafenib, sorafenib exhibited antagonistic effects dependent on AKT, suggesting a disadvantage of sorafenib in advanced CRC [98]. Another study confirmed that sorafenib monotherapy was inactive against patient-derived CRC cells; however, its combination with the MEK inhibitor selumetinib exhibited distinct antiproliferative activity, which was superior to the activity of cetuximab plus selumetinib. Synergistic effects of sorafenib plus selumetinib were observed in cells with the MAP2K1 K57T mutation responsible for EGFR inhibitor resistance [99]. Sorafenib binds to an inactive BRAF conformation (which differs from the vemurafenib binding conformation), leading to reduced BRAF interactions with distinct proteins upon sorafenib binding [100]. It was shown that sorafenib treatment led to ARAF homodimer and ARAF/BRAF heterodimer (together with the scaffold protein KSR1) formation, which induced MAPK signaling and tumor invasiveness (Figure 1b) [101]. In addition, the pronounced long-term activity of sorafenib in a lung adenocarcinoma patient was based on an ARAF mutation, which sensitized the tumor to sorafenib therapy [102]. Promising effects of sorafenib were observed in a vemurafenib-insensitive melanoma cell line with activated MAPK signaling based on a BRAF fusion protein without known oncogenic mutations. Sorafenib was distinctly more active than vemurafenib against these cells and efficiently blocked MAPK activation [103].
The search for further mutations in vemurafenib-resistant BRAFV600E mutant A431 melanoma cells identified an in-frame deletion in the BRAFV600E locus as well as a point mutation in the transcription repressor BCORL1 in the resistant cells. Sorafenib could re-sensitize these resistant cells to vemurafenib since it is not affected by these new mutations [104]. In addition, sorafenib showed promising activity against non-V600 BRAF mutant cancers. BRAF-G469R and BRAF-N581S mutant lung cancer cells revealed considerable sensitivity to sorafenib treatment [105]. The combination of sorafenib with the MEK inhibitor selumetinib showed synergy in non-V600 tumor cell lines, including MDA-MB-231 triple-negative breast carcinoma (G464V), based on increased apoptosis induction and ERK1/2 inhibition, and strongly inhibited MDA-MB-231 xenograft growth in mice [106]. Sorafenib also exhibited strong synergistic effects on MDA-MB-231 cells in combination with the CDK inhibitor flavopiridol, which was associated with apoptosis induction and downregulation of Rb (retinoblastoma) and MCL1 [107]. Analogously to sorafenib, the VEGFR/BRAF inhibitor regorafenib showed activity against BRAFV600E- and non-V600 mutant thyroid cancers, unlike the V600E-specific inhibitor dabrafenib [108]. It is noteworthy that sorafenib sensitized TNF-α-related apoptosis-inducing ligand (TRAIL)-resistant (BRAFV600E) HT-29 CRC to TRAIL-induced cell death in vitro and in vivo via suppression of NF-κB, MCL-1, and phospho-MEK/ERK1/2 in particular since TRAIL-resistance was described as a resistance mechanism of the BRAF inhibitor PLX-4720 based on oncogenic PIK3CA (Figure 1b) [109][110]. Finally, the combination of sorafenib with EGFR inhibitors (erlotinib or cetuximab) led to synergistic effects in CRC and NSCLC cells based on downregulated MAPK, AKT, and VEGFR signaling [111].
Various other multikinase inhibitors with VEGFR-inhibitory activity were studied in BRAF mutant cancers. Resistance to sunitinib was associated with the upregulation of RAS/MAPK signaling in BRAF mutant thyroid cancers, as well as the stimulation of wild-type cells with EGF, which occurred independently from the expression of VEGFR1-3 and other RTKs such as PDGFRA and KIT [112]. Pazopanib inhibits MAPK signaling in BRAF mutant HER2-positive breast cancer cells. Phosphorylation of MEK1/2, ERK1/2, VEGFR1, and VEGFR2 was reduced in pazopanib-treated HER2-transfected MDA-MB-231 tumor cells and brain metastases. While no interaction with BRAFV600E was found, pazobanib was able to interact with the exon 11 oncogenic BRAF mutant [113].
Recently, the combination of axitinib with the BRAF inhibitors dabrafenib or PLX4720 exhibited an additive effect on BRAFV600E mutant anaplastic thyroid carcinoma (ATC) cells, and blocked cell invasion and migration. Axitinib induced c-Jun signaling and downregulated histone H3 and aurora kinase (AURKA) phosphorylation. In an orthotopic ATC mouse model, axitinib plus a BRAF inhibitor inhibited tumor growth and led to prolonged survival [114]. In addition, the combination of axitinib with vemurafenib inhibited tumor growth of BRAFV600E HT-29 and COLO-205 xenografts by suppressing cytokines and growth factor release (MIF, IL8, TGF-β, and VEGF-A) (Figure 1b) [115]. Ponatinib plus PLX4720 revealed synergistic activity against BRAFV600E ATC cells. The combined treatment induced apoptosis, suppressed MEK and ERK phosphorylation as well as c-Jun signaling, and blocked ATC colony formation and migration (Figure 1b). This combination therapy also re-sensitized PLX4720-resistant BRAFV600E cells and inhibited tumor growth in orthotopic ATC mouse models [116]. Admittedly, the described multikinase inhibitors with VEGFR inhibitory activity may also exert their effects on BRAF mutant cancers in part by targeting other RTKs. Such effects are mentioned in the sections that follow about the respective RTKs, e.g., in the section about PDGFR/KIT for sunitinib and regorafenib.
The development of new dual VEGFR/BRAF inhibitors by combining essential VEGFR- and BRAF-inhibitory molecular scaffolds has also shown promising results, and the dual VEGFR/BRAF inhibitor RAF265 has already reached clinical trials. RAF265 is an optimized molecule derived from the chemical combination of the crucial structural motifs of sorafenib and pazopanib [92]. RAF265 exhibited strong antiproliferative activities against HT-29 (BRAFV600E) and MDA-MB-231 (BRAFG463V) tumor cells and inhibited MDA-MB-231 xenograft growth in vivo [117]. Another in vivo study described the tumor growth inhibitory activity of RAF265 in HT-29 xenografts, as well as decreased MAPK signaling in mutant CRC cells even in the presence of EGFR-activating EGF [118]. In addition, RAF265 was active against patient-derived advanced melanomas, which were orthotopically implanted into mice, but only 29% of the responding melanomas were BRAFV600E/K mutant, while all others were wild-type melanomas [119]. RAF265 in combination with the PI3K inhibitor ZSTK474 showed synergistic effects in thyroid carcinoma cells, including BRAF mutant cell lines [120]. A new tyrphostin derivative with VEGFR2 inhibitory activity was particularly antiproliferative against BRAFV600E CRC cell lines (HT-29 and COLO-201) and showed pro-apoptotic and anti-migratory activities [121].
2.2. Additional Mechanisms and Factors of VEGFR-Mediated BRAF Inhibitor Resistance
The BRAF inhibitor dabrafenib reduced VEGF-A release and downregulated VEGFR2 expression in sensitive A431 melanoma cells. However, in A431R cells with acquired dabrafenib resistance, dabrafenib increased VEGF-A secretion without changing VEGFR2 levels, leading to enhanced invasiveness. The PI3K/mTOR inhibitor GSK2126458A blocked dabrafenib-induced VEGF-A release and invasiveness in the resistant cells, thus indicating AKT/mTOR signaling as a suitable target in BRAF inhibitor-resistant cancers associated with increased VEGFR2 activity. In addition, bevacizumab inhibited the pro-invasive properties of dabrafenib in these cells [122]. Vemurafenib-treated BRAF mutant thyroid cancer cells also showed an upregulation of VEGFR2 associated with SHP2 activation [47]. Suppression of miR-126-3p also contributed to dabrafenib resistance in BRAF mutant melanoma by upregulating VEGF-A and ADAM9 (a disintegrin and metalloproteinase domain 9) (Figure 1b) [123]. The antidiabetic drug metformin was described as a possible anticancer agent against various cancers by activating AMPK (AMP-activated protein kinase). However, in BRAFV600E mutant melanoma, metformin upregulated VEGF-A and enhanced angiogenesis and tumor growth both in vitro and in vivo. Mechanistically, metformin-activated AMPK suppresses the phosphatase DUSP6, which is accompanied by ERK activation and VEGF-A release (Figure 1b). In addition, the protein kinase RSK was activated in the metformin-resistant cells, leading to sustained TORC1 signaling. Only in the case of VEGF signaling blockade was metformin able to suppress tumor growth [124].
FAK (focal adhesion kinase) forms complexes with VEGFR3 to promote invasiveness and angiogenesis and can be targeted with FAK inhibitors. Treatment of BRAF mutant melanoma cells with the FAK inhibitor chloropyramine disrupted FAK–VEGFR3 complex formation and suppressed phosphorylation of FAK and VEGFR3 associated with inactivation of ERK1/2 (Figure 1b) [125]. The establishment of BRAFV600E mutation in melanomas was associated with upregulated WNT5A expression leading to increased VEGF secretion and vascularization of melanomas, which can explain the high susceptibility of BRAFV600E melanomas to bevacizumab treatment in a clinical phase III trial. WNT5A is a ligand of RTK ROR2 (receptor tyrosine kinase-like orphan receptor 2), which is upregulated by MAPK signaling in BRAFV600E cells (Figure 1b) [126]. In addition, the combination of the Wnt/β-catenin inhibitor pyrvinium with vemurafenib inhibited tumor growth in BRAFV600E HT-29 and COLO-205 CRC xenografts. Pyrvinium plus vemurafenib blocked the secretion of cytokines and growth factors (MIF, IL8, TGF-β, and VEGF-A) [115].
2.3. Clinical Trials of VEGFR Inhibitors in BRAF Mutant Cancers
Several VEGFR inhibitors, such as sorafenib and becacizumab, are already approved for the therapy of several cancer diseases, which simplified the access of these approved drugs for new clinical trials in combination with BRAF and/or MEK inhibitors. Various clinical trials of VEGFR-inhibitory drugs for the treatment of BRAF mutant cancers disclosed promising results. While sorafenib monotherapy showed limited activity in BRAFV600E mutant melanoma patients, another early study of carboplatin and paclitaxel chemotherapy combined with sorafenib in melanoma patients suggested a positive effect of sorafenib in NRAS mutant melanoma patients [127][128]. However, grade III/IV toxicities were higher in the sorafenib-treated groups. A case report from 2013 described the treatment history of a BRAFV600E mutant CRC patient suffering from progressive disease upon treatment with FOLFOX followed by FOLFIRI plus cetuximab. Off-label treatment of this patient with sorafenib and cetuximab led to stable disease for seven months, followed by treatment with regorafenib plus panitumumab, which slowed down disease progression considerably [129]. The combination of vemurafenib with sorafenib (VS) was well-tolerated in 24 BRAF mutant cancer patients (including melanoma and other cancers) and displayed a partial response in five patients, including melanoma, lung adenocarcinoma, papillary thyroid cancer, and two ovarian cancer patients. All responding patients had the BRAFV600E mutation. The two ovarian cancer responders are especially interesting because they were pre-treated with a MEK inhibitor or a combination of BRAF, MEK, and ERK inhibitors, which suggests a promising outcome of the vs. therapy in future clinical trials with pre-treated BRAFV600E mutant ovarian cancer [130]. In terms of metastatic CRC, regorafenib is clinically applied as a therapy for pre-treated patients with progressive disease after two phase 3 clinical trials (CORRECT and CONCUR studies) from 2013 and 2015 revealed significantly prolonged overall survival [131][132]. Notably, regorafenib also showed activity as a second-line therapy in patients with advanced CRC pre-treated with bevacizumab plus trifluridine/tipiracil [133]. Meanwhile, the dual RAF/VEGFR2 inhibitor RAF265 has undergone a first-in-human phase 1 clinical trial with melanoma patients. Four partial responses were observed in patients with the BRAFV600E mutation, three partial responses, and one complete response in BRAF-wild-type melanoma patients. A significant decrease in VEGFR-2 levels was observed in the RAF265-treated patients [134].
Bevacizumab is applied as a first-line therapy for advanced CRC in combination with chemotherapy (with FOLFOX/FOLFIRI or with FOLFOXIRI) and showed a significantly better outcome in non-V600 BRAF mutant CRC patients (median overall survival of 38.0 months) than in V600E mutant CRC (median overall survival of 18.2 months) [135]. A recently published phase 2 study (FIRE-4.5) of FOLFOXIRI plus cetuximab or bevacizumab in BRAFV600E CRC patients underlined the superior activity of bevacizumab when compared with cetuximab and its preferred application as first-line therapy for this cancer disease [136]. A pooled analysis of anti-angiogenic biologicals, including bevacizumab, ramucirumab, and aflibercept, confirmed the efficacy of these drugs as a second-line treatment of pre-treated BRAF mutant CRC [137]. In addition, a retrospective analysis of the maintenance therapy of FOLFOX-pre-treated BRAFV600E metastatic CRC with bevacizumab plus fluoropyrimidine showed high disease control (74%), and overall survival (25.6 months), warranting further large-scale trials of this promising second-line combination therapy in the future [138]. The global phase 3 RAISE trial with metastatic CRC patients pre-treated with bevacizumab, oxaliplatin, and fluoropyrimidine showed that the anti-VEGFR2 antibody ramucirumab plus FOLFIRI prolonged overall survival and progression-free survival together with an increase of side effects (neutropenia, thrombocytopenia, stomatitis, epistaxis, and hypertension) when compared with FOLFIRI plus placebo, which was accompanied by a substantial benefit of ramucirumab plus FOLFIRI in BRAF mutant patient groups [139]. The recombinant fusion protein aflibercept inhibits VEGF and PlGF, which makes it an interesting alternative for bevacizumab-resistant cancers, since bevacizumab only inhibits VEGF, and PlGF was identified as a bevacizumab resistance factor. The phase 3 VELOUR trial with aflibercept plus FOLFIRI for the treatment of oxaliplatin-pretreated metastatic CRC patients showed anti-VEGF-associated adverse effects together with casually increased chemotherapy-associated toxicities and improved overall and progression-free survival when compared with FOLFIRI plus placebo as second-line therapy. In particular, the aflibercept-treated BRAF mutant patient subgroup of the VELOUR trial exhibited a doubled median survival time (10.3 months) when compared with placebo-treated patients (5.5 months) [140]. A case report with aflibercept plus FOLFIRI as a follow-up therapy for a metastatic BRAFV600E mutant CRC patient after bevacizumab plus FOLFIRI and targeted BRAF therapies showed a pronounced response and stable disease for more than four months (ongoing). Another metastatic CRC patient with the BRAFD594N mutation, who was pre-treated with bevacizumab, ramucirumab, and cetuximab, experienced a progression-free survival of approximately one year after being treated with aflibercept plus FOLFIRI [141].
The effects and mechanisms of the described VEGFR inhibitors are summarized in Table 2.
Table 2. VEGFR inhibitors and their activities in BRAF mutant cancers.
| VEGFR Inhibitor | Mechanisms (Cancers/Cell Lines) | Clinical Studies | References |
|---|---|---|---|
| Sorafenib | Superior to vemurafenib (BRAFV595E canine transitional cell carcinoma cells); apoptosis induction; suppression of MAPK and AKT; reduced p-VEGFR1/2/3 (BRAFV600E thyroid carcinoma cells); active against vemurafenib-insensitive melanoma cell cells with BRAF fusion protein, drug resensitization (vemurafenib-resistant BRAFV600E mutant A431 melanoma cells with BRAFV600E loci and BCORL1 mutations); active against non-V600 mutation (BRAF-G469R and BRAF-N581S mutant lung cancer cells); synergy in combination with the MEK inhibitor selumetinib or the CDK inhibitor flavopiridol in non-V600 tumors (MDA-MB-231 triple-negative breast carcinoma (G464V)); sensitization to TRAIL (BRAFV600E HT-29 CRC) | Stable disease for seven months in combination with cetuximab (case report, pretreated BRAFV600E mutant CRC patient); partial response in combination with vemurafenib (BRAFV600E mutant melanoma, lung adenocarcinoma, papillary thyroid cancer, and ovarian cancer) | [96][97][103][104][105][106][107][109][129][130] |
| Regorafenib | Activity against BRAFV600E and non-V600 mutant thyroid cancers | Combination with panitumumab slowed down disease progression (case report, pretreated BRAFV600E mutant CRC patient) | [108][129] |
| Sunitinib | Sunitinib resistance by upregulation of RAS/MAPK (BRAF mutant thyroid cancers) | - | [112] |
| Pazopanib | Blocking of MAPK signaling (BRAF mutant HER2-positive breast cancer cells); reduced p-MEK1/2, p-ERK1/2, and p-VEGFR1/2 (HER2-transfected MDA-MB-231 tumor cells and brain metastases); no interaction with BRAFV600E but with exon 11 BRAF mutant | - | [113] |
| Axitinib | Additive effect in combination with dabrafenib or PLX4720; blocking of invasion and migration; induction of c-Jun signaling and suppression of p-H3 and p-AURKA (BRAFV600E mutant ATC); tumor growth inhibition and prolonged survival (orthotopic ATC mouse model); combination with vemurafenib suppressed tumor growth; cytokine and growth factor release (BRAFV600E HT-29 and COLO-205 CRC xenografts) | - | [114][115] |
| Ponatinib | Synergistic activity in combination with PLX4720; pro-apoptotic; suppression of p-MEK/ERK and c-Jun (BRAFV600E ATC cells); re-sensitization to PLX4720 (PLX4720-resistant BRAFV600E ATC); inhibition of tumor growth (orthotopic ATC mouse models) | - | [116] |
| RAF265 | Strong antiproliferative activities (BRAFV600E HT-29 and BRAFG463V MDA-MB-231 cells); inhibition of tumor growth (HT-29 and MDA-MB-231 xenografts); active against patient-derived advanced melanomas; synergistic effects in combination with PI3K inhibitor ZSTK474 (thyroid carcinoma cells including BRAF mutant cell lines) | Four partial responses and significant decrease in VEGFR-2 levels (first-in-human phase 1 clinical trial, melanoma with the BRAFV600E mutation) | [117][118][119][120][134] |
| Bevacizumab | Inhibition of pro-invasive properties of dabrafenib (dabrafenib-resistant A431R melanoma cells) | Clinically applied as first-line therapy of RAS and/or BRAF mutant CRC; promising second-line combination therapy plus fluoropyrimidine with high disease control (74%) and overall survival (25.6 months, FOLFOX pre-treated BRAFV600E metastatic CRC) | [95][122][135][136][138] |
| Ramucirumab | - | Efficacy as a second-line treatment of pre-treated BRAF mutant CRC; substantial benefit plus FOLFIRI in BRAF mutant patient groups (global phase 3 RAISE trial; metastatic CRC patients pre-treated with bevacizumab, oxaliplatin, and fluoropyrimidine) | [137][139] |
| Aflibercept | Inhibition of VEGF and PlGF and breach of bevacizumab resistance | Efficacy as a second-line treatment of pre-treated BRAF mutant CRC; improved overall and progression-free survival in combination with FOLFIRI (phase 3 VELOUR trial, oxaliplatin-pretreated metastatic CRC); pronounced response and stable disease for more than four months as follow-up therapy in combination with FOLFIRI (case report, pretreated metastatic BRAFV600E mutant CRC); progression-free survival (ca. one year) in combination with FOLFIRI (case report, pretreated metastatic BRAFD594N CRC) | [137][140][141] |
3. PDGFR and KIT in BRAF Mutant Cancers
3.1. PDGFRs and KIT Receptors, Inhibitors, and Mechanisms
The platelet-derived growth factor receptors PDGFRA and PDGFRB (PDGFR-α and PDGFR-β, respectively) play an important role in cancer progression. Both receptors are induced by the binding of PDGF ligands such as PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD [142]. Upon ligand binding, all three possible PDGFR dimers, the homodimers PDGFRAA and PDGFRBB, and the heterodimer PDGFRBB, can be formed and activated by transphosphorylation. Ras/MAPK, PI3K/AKT, and PLCγ signaling are activated by PDGFR to transmit mitogenic signals [143].
Overexpression of PDGFR was correlated with shorter overall survival and co-amplification with other RTKs in lung cancer, ovarian cancer, and medulloblastomas [144][145][146]. PDGFRA mRNA overexpression was associated with metastasis and shorter patient survival times in oral squamous cell carcinoma [147][148]. In addition to overexpression, various PDGFRA mutations are of relevance for tumor formation and progression [149]. PDGFRA mutations of the regulatory domains (extracellular domain and juxtamembrane domain) or of the tyrosine kinase domain eventually cause ligand-independent receptor dimerization or kinase activation independent from receptor dimerization [150]. For example, PDGFRA mutations in exon 18 were often described in gastrointestinal stromal tumors (GISTs) [151][152][153]. In addition, the V546E gain-of-function mutation promotes ERK and STAT5 phosphorylation [154]. In hematological cancers, gene fusions of PDGFR are often identified. The FIP1L1-PDGFRA fusion protein shows upregulated kinase function [155]. In terms of PDGFRB fusions, most protein partners promote dimerization and kinase activation in myeloid neoplasms as well [156][157]. Various solid tumors such as thyroid, brain, breast, and ovarian cancers strongly depend on autocrine PDGFR activation followed by induction of MAPK, PI3K/AKT, and STAT signaling [158][159][160][161].
The KIT receptor is a stem cell factor (SCF) receptor with high homology with PDGFRs, which regulates stem cell maintenance and differentiation under normal circumstances [162]. However, deregulated KIT activity leads to various cancer diseases. KIT-activating mutations were observed in the majority of GISTs [163]. KIT activation was also described as a pro-apoptotic factor in melanoma, and increased KIT mRNA levels were found in BRAFV600 mutant melanoma patients who responded well to combined BRAF inhibitor and MEK inhibitor treatment [164][165]. Loss-of-function mutations of KIT were responsible for melanoma, thyroid, and breast cancers [166][167][168]. KIT directly activates downstream signaling pathways such as Ras/MAPK, PI3K/AKT, and Src signaling, which can be blocked by KIT inhibitors, including the already-described multikinase inhibitors sorafenib and regorafenib, as well as imatinib (an ABL kinase inhibitor that also targets KIT and PDGFR) [169].
In A375 melanoma with BRAFV600E mutation, PLX4720 treatment showed reduced efficacy; however, the antitumor activity was restored by treatment with the PDGFR inhibitor imatinib, and imatinib in combination with BRAF inhibitors suppressed both phospho-AKT and phospho-ERK [170]. The synergistic effects of the PDGFR/KIT inhibitor masatinib in combination with the BRAF inhibitor PLX4720 can also occur independently from AKT signaling and MAPK reactivation in BRAFV600E melanoma [21].
BRAFV600E mutant melanomas acquire vemurafenib resistance by redundant activation of MAPK signaling via PDGFRB [171]. The concomitant activation of MAPK and AKT signaling by upregulated PDGFRB upon treatment with vemurafenib in BRAFV600E melanoma cells was overcome by combinatory inhibition of BRAF, PI3K, and mTORC1/2 [172]. In addition, PDGFRB signaling was described as a MEK inhibitor therapy escape mechanism in melanoma, yet Hsp90 plays a substantial role as an RTK regulator in these drug resistance mechanisms. The Hsp90 inhibitor XL888, which suppressed RTK expression, in combination with the PDGFR inhibitor crenolanib exhibited promising activities against BRAF inhibitor-resistant melanoma cells (Figure 1c) [173]. Functional PDGFRB proteins can also be transferred to BRAFV600E melanoma cell recipients by extracellular vesicles derived from PLX4720-resistant cells, which is accompanied by PI3K/AKT activation and BRAF inhibitor resistance formation in the receiving cells [174]. The AKT-dependent adaptive response in BRAF mutant melanoma was associated with PDGFRB upregulation. While PTEN expression limited this response, the AKT1 (Q79K) mutant restored PTEN-suppressed PI3K/AKT pathway and enhanced BRAF inhibitor-dependent AKT signaling and resistance [175]. PDGFRB induction was associated with ERK5 activation in NRAS mutant melanoma, but MEK/ERK5 co-targeting (trametinib plus ERK5 inhibitor XMD8-92) is also of relevance for BRAF mutant cancers since it suppressed tumor growth of the BRAFV600E mutant LOX-IMVI melanoma cell line (Figure 1c) [176]. It is worth mentioning that the multikinase inhibitor sorafenib inhibited wild-type and gatekeeper mutant PDGFRB associated with imatinib resistance and suppressed phospho-PDGFRB in Ba/F3 hybridoma cells [177]. The BET (bromodomain and extra-terminal domain) inhibitor PLX51107 blocked adaptive BRAF and MEK inhibitor-mediated upregulation of ErbB3 and PDGFRB in BRAFV600E melanomas both in vitro and in vivo and increased the anticancer activity of BRAF and MEK inhibitors, which indicates the eminent role of bromodomain epigenetic reader proteins such as BRD2/4 as enhancers of RTK upregulation in BRAF mutant melanoma [178]. Suppression of SOX10-induced TGF-β signaling is followed by upregulation of PDGFRB in BRAF inhibitor-resistant melanomas (Figure 1c) [56].
The activation of PDGFRA, accompanied by upregulated MAPK and AKT signaling, was induced in BRAFV600E melanoma resistant to vemurafenib. The Shh (sonic hedgehog) pathway and Gli activation were associated with PDGFRA-mediated vemurafenib resistance, and the Shh inhibitor LDE225 was able to circumvent this vemurafenib resistance mechanism (Figure 1c). In addition, the PDGFR inhibitors sunitinib and crenolanib showed pro-apoptotic effects on BRAF inhibitor-sensitive and -resistant BRAFV600E melanoma cells [179]. PDGFRA upregulation was also observed upon SHP2 activation in vemurafenib-resistant thyroid cancer cells (Figure 1c) [47].
The new and selective PDGFR inhibitor AG1296, a tyrphostin derivative, suppressed phosphorylation of PDGFRA and PDGFRB in vemurafenib-resistant BRAFV600E A375 melanoma cells, which was associated with apoptosis induction and blocked cell migration. Moreover, AG1296 strongly inhibited in vivo tumor growth of the resistant A375 melanoma model, and this drug candidate can apparently become a valuable treatment option for BRAF mutant vemurafenib-resistant melanoma [180]. AG1296 also exhibited promising anticancer activity against ATC in combination with the AKT inhibitor MK-2206, and further studies about this drug combination will show if it can become a new milestone for ATC therapy [181]. The VEGFR/PDGFR inhibitor cediranib suppressed PDGFRA/B phosphorylation in PLX4720-resistant BRAFV600E melanoma cells and revealed synergistic effects in combination with PLX4720 and selumetinib in these resistant cells [182].
In terms of the KIT receptor, the suppression of KIT expression in BRAFV600E melanoma cells by G-quadruplex ligands, which target the Kit gene, downregulated MAPK and AKT signaling at the transcription and post-translational stages [183]. The activation of the transcription factor MYC plays a crucial role in the formation of intrinsic and acquired BRAF/MEK inhibitor resistance in BRAF mutant melanoma (Figure 1c). MYC is located downstream of resistance initiation mechanisms such as MAPK reactivation and the activation of PI3K/AKT, Notch, and other signaling pathways. MYC-driven resistant melanomas were especially vulnerable to treatment with KIT and Src family kinase inhibitors, which indicates the important role of KIT signaling in the development of MYC-mediated BRAF inhibitor resistance and suggests KIT as a suitable drug target [184]. The protein SPRED1 (Sprouty-related Ena/VASP homology [EVH1] domain containing 1) is a tumor suppressor, in particular, in KIT mutant melanoma, which downregulates MAPK signaling. Yet, suppressed SPRED1 lowered dabrafenib efficacy and promoted dabrafenib resistance in BRAF mutant melanoma due to sustained MAPK activation and KIT signaling. It was shown that SPRED1 is located downstream of KIT and interacts with NF1, thus forming a direct inhibitor of Ras (Figure 1c) [185]. KIT mutations occur frequently in GISTs, leading to tumors that are resistant to imatinib treatment. However, imatinib resistance of Ba/F3 KITT670I mutant hybridoma cells can be managed by treatment with the multikinase (RAF/RTK) inhibitor sorafenib [186]. In imatinib-resistant BRAF mutant GIST, imatinib could suppress mutant KIT activity, but it was not able to suppress ERK1/2 activation or MAPK signaling [187]. Imatinib is clinically applied for the off-label treatment of KIT mutant melanoma (approximately 1% of all melanomas), and, thus, the increasing knowledge of KIT inhibitors can lead to beneficial therapies for drug-resistant BRAF mutant cancers, including melanomas [188]. Ripretinib was shown to inhibit all KIT and PDGFR activation loop mutants by inhibition of the activation loop switch and suppress KIT signaling together with ERK, AKT, and STAT5 in an imatinib-resistant patient-derived KIT mutant GIST xenograft model [189].
3.2. Clinical Trials of PDGFR and KIT Inhibitors in BRAF Mutant Cancers
As already mentioned above, the multikinase inhibitor sorafenib, which also targets PDGFR and KIT, showed promising results in NRAS mutant melanoma patients in combination with carboplatin and paclitaxel [128]. Imatinib is the drug of choice for the treatment of KIT mutant GIST; however, BRAF mutant GISTs cannot be treated with imatinib because of its lack of efficacy [190]. Sorafenib was active in patients with imatinib-, sunitinib-, or regorafenib-resistant BRAF-wild-type GIST, but it was inactive in a BRAFV600E GIST patient [191]. The PDGFR/KIT inhibitor ripretinib underwent a phase 3 trial (INTRIGUE) in comparison with the multikinase inhibitor sunitinib in imatinib-pretreated GIST patients, and although ripretinib was not superior to sunitinib in terms of survival, it showed reduced side effects accompanied by a better quality of life when compared with sunitinib [192]. Yet, it remains to be elucidated if ripretinib also works in BRAF mutant cancers because of its low BRAF and CRAF inhibitory activity [189]. A case report of a GIST patient with the BRAFV600E mutation showed progression upon treatment with imatinib, sunitinib, and a VEGFR/PDGFR/KIT inhibitor (as part of a clinical study) but exhibited tumor regression by dabrafenib treatment before the tumor progressed after 8 months due to a PIK3CA (H1047R) gain-of-function mutation leading to BRAF inhibitor resistance [193]. Regorafenib was approved for the third-line therapy of KIT mutant advanced GIST after imatinib/sunitinib failure following the successful phase 3 GRID trial [194]. The case report of a BRAFV600E mutant KIT/PDGFRA-wild-type metastatic GIST patient showed a complete response to first-line regorafenib treatment [195]. The phase 2 REGISTRI trial with regorafenib as a first-line treatment for KIT/PDGFRA-wild-type metastatic GIST patients revealed promising results, which suggest the consideration of regorafenib as a suitable first-line therapy for the rare KIT/PDGFRA-wild-type GIST. A BRAFV600E mutant succinate dehydrogenase (SDH)-proficient GIST patient in this study experienced a progression-free survival of 3.45 months [196].
Table 3 summarizes the effects and mechanisms of the described PDGFR and KIT inhibitors.
Table 3. PDGFR and KIT inhibitors and their activities in BRAF mutant cancers.
| PDGFR/KIT Inhibitor | Mechanisms (Cancers/Cell Lines) | Clinical Studies | References |
|---|---|---|---|
| Imatinib | Suppression of p-AKT and p-ERK in combination with BRAF inhibitors; restoration of PLX4720 activity (animals with BRAFV600E A375 melanoma); suppression of mutant KIT activity but no inhibition of MAPK signaling (imatinib-resistant BRAF mutant GIST) | Inactive against BRAF mutant GIST | [170][187][190][193] |
| Masatinib | Synergy in combination with BRAF inhibitor PLX4720 independent from AKT and MAPK reactivation (BRAFV600E melanoma) | - | [21] |
| Crenolanib | Active in combination with Hsp90 inhibitor XL888 (BRAF inhibitor-resistant melanoma cells); pro-apoptotic effects (BRAF inhibitor-sensitive and -resistant BRAFV600E melanoma cells) | - | [173][179] |
| Sunitinib | Pro-apoptotic effects (BRAF inhibitor-sensitive and -resistant BRAFV600E melanoma) cells | Inactive against BRAF mutant GIST | [179][193] |
| AG1296 | Suppression of p-PDGFRA and p-PDGFRB; apoptosis induction and inhibition of cell migration; inhibition of tumor growth in vivo (vemurafenib-resistant BRAFV600E A375 melanoma) | - | [180] |
| Cediranib | Suppression of p-PDGFRA/B; synergistic effects in combination with PLX4720 and selumetinib (PLX4720-resistant BRAFV600E melanoma cells) | - | [182] |
| Regorafenib | - | Complete response by first-line treatment (case report, BRAFV600E mutant KIT/PDGFRA-wild-type metastatic GIST); progression-free survival of 3.45 months of a BRAFV600E mutant succinate dehydrogenase (SDH)-proficient GIST patient (phase 2 REGISTRI trial with regorafenib as a first-line treatment for KIT/PDGFRA-wild-type metastatic GIST) | [195][196] |
4. FGFR in BRAF Mutant Cancers
There are four known fibroblast growth factor receptors (FGFR1-4), which bind to 22 different FGFs. Only 18 FGFs can induce FGFR dimerization and activation upon binding to the FGFR extracellular ligand binding domain [197]. FGFR activation leads to upregulation of the MAPK, PI3K/AKT, STAT, and PLCγ/PKC signaling pathways. Overexpression of FGFs and FGFRs, activating FGFR mutations, and fusion proteins were described in FGFR-dependent cancer progression and oncogenic signaling induction associated with cancer drug resistance [198].
In addition to BRAF mutations, melanoma often displays overexpression of FGFs, including FGF2 (also known as basic FGF/bFGF), FGF5, and FGF18. The FGFR inhibitors SU5402 and PD166866 showed antiproliferative and pro-apoptotic effects on BRAFV600E melanomas and synergistically augmented the anticancer activities of sorafenib and the BRAFV600E-selective inhibitor RG7204, in association with suppressed AKT5 and STAT3 phosphorylation [199]. Upregulation of FGF1 secretion was observed in BRAFV600E mutant melanoma resilience upon vemurafenib treatment, which was accompanied by reduced pro-apoptotic activity of vemurafenib and activation of HGF expression by fibroblasts. FGF1 was upregulated in the vemurafenib-resistant cells by PI3K/AKT signaling as well as by suppression of FRA1, which is a component of the transcription factor AP-1 (Figure 1d) [200]. Increased FGF5 expression in BRAFV600E melanomas led to increased MAPK and NFAT (nuclear factor of activated T-cells) signaling (without effects on STAT3), tumor growth, and anti-apoptotic effects (Figure 1d) [201]. FGFs are regulated by proteinases, and activation of FGF2 signaling, including MAPK upregulation, was blocked in breast cancer cells by the transmembrane proteinase MT1-MMP (membrane-type 1 matrix metalloproteinase, MMP-14). But instead of proteolytic cleavage of FGF2, MT1-MMP suppressed FGF2 signaling by degradation of the FGF2-binding receptors FGFR1 and FGFR4 [202]. In wild-type melanoma cells, FGFR activation led to increased proliferation and ERK1/2 activation, while BRAFV600E cells showed no changes since they already possess a high ERK1/2 activation level, and the antiproliferative activity of FGFR inhibitors (ponatinib, BGJ-398, BIBF-1120, and AZD-4547) was observed independent from the mutation status of treated melanoma cells [203]. Yet, the acquired resistance of BRAFV600E melanoma to vemurafenib treatment was associated with upregulated FGFR3/Ras signaling (increased phospho-FGFR3, phospho-MEK, and phospho-ERK1/2 levels), leading to vemurafenib-irresponsive MAPK reactivation, which can be blocked by a combination of MEK and pan-RAF inhibitors (selumetinib and RAF265) [204]. FGFR1 and FGFR3 are highly upregulated RTKs in CRC cell lines, and the multi-target angiokinase inhibitor dovitinib suppressed KRAS mutant LoVo and BRAFV600E mutant HT-29 xenograft growth and downregulated phospho-FGFR1 and phospho-ERK levels in both xenografts [205]. Vemurafenib-resistant RKO CRC cells possessed activated phospho-FGFR, but dovitinib sensitized RKO cells to vemurafenib. Dovitinib also augmented the activity of vemurafenib and selumetinib in the BRAF- and MEK inhibitor-resistant BRAF mutant YUKSI melanoma cells and in the vemurafenib-resistant YUMAC XR4MC8 melanoma xenograft model [206]. A diet containing aspirin (acetylsalicylic acid) reduced the size and number of distant colorectal cancer metastases in BrafV637E/+ and Villin-CreERT2/+ mice associated with suppression of FGFR, PI3K, and Notch signaling, while tumors in untreated mice showed activation of FGFR and PI3K signaling [207].
The phosphatase SHP2 upregulated FGFR1-4 in BRAFV600E thyroid cancers, which led to MAPK reactivation and vemurafenib resistance [47]. Increased unfolded protein response (UPR) by upregulated ATF6, PERK, and IRE1 was observed in BRAFV600E patient-derived metastatic melanoma when compared with non-metastatic cells, and active UPR also induced FGF1 and FGF2 expression in the metastatic cells (Figure 1d). Treatment of the metastatic melanoma cells with the UPR antagonist 4-phenylbutyric acid downregulated FGF expression and cell motility [208]. In terms of non-V600 BRAF mutant cancers, feedback activation of FGFR occurred upon MEK inhibition in non-V600E mutant lung adenocarcinoma cells, and FGFR activated MAPK signaling in lung cancers with non-V600E mutations [30].
The multikinase inhibitors nintedanib, dovitinib, and lucitanib have undergone clinical trials for the treatment of various advanced pre-treated cancers with FGFR alterations. However, there has neither been an approval nor an investigation of BRAF mutant patient status in relation to drug response. In addition to multikinase inhibitors acting on FGFRs, two selective FGFR inhibitors were already approved for the treatment of FGFR3-altered urothelial cancer (erdafitinib) and FGFR2-altered cholangiocarcinoma (pemigatinib), while other selective FGFR inhibitors are in clinical trials [197]. The second-generation FGFR-selective inhibitors AZD-4547 and BGJ-398 were developed to reduce the off-target side effects of the multikinase inhibitors and showed promising results from phase 1/2 studies with FGFR-altered advanced cancers [209]. Notably, BGJ-398 is currently being investigated in a recently completed LOGIC-2 phase 2 trial with patients suffering from BRAF/MEK inhibitor-resistant advanced or metastatic BRAFV600E melanoma in order to evaluate the potential of BGJ-398 to treat this highly problematic melanoma type [210].
5. MET Kinase/HGFR in BRAF Mutant Cancers
The MET (c-Met) kinase, also known as hepatocyte growth factor (HGFR), is an RTK that is activated upon binding to the hepatocyte growth factor (HGF) [211]. MET and HGF are overexpressed in various cancers. Associated with the MET exon 14 mutation, metastasis formation in advanced NSCLC (via RhoA overexpression) and breast cancer patients was observed [212][213][214]. Unregulated dimerization of MET fusions with functional MET kinase domains is a consequence of MET exon 15 fusion proteins (e.g., HLA-DRB1-MET), which can be tackled by MET inhibitors (multikinase MET inhibitors crizotinib, cabozantinib, and MET-selective inhibitor tepotinib) [215]. MET fusion proteins were reported in lung cancer, e.g., HLA-DRB1-MET fusion, and the MET-UBE2H fusion protein was discussed as a factor responsible for EGFR inhibitor (erlotinib) resistance in a case report, which was successfully treated with the MET inhibitor crizotinib [216][217]. Autocrine HGF/MET signaling contributed to sorafenib resistance in hepatocellular carcinoma (HCC), since resistant HCC cells exhibited increased HGF levels and MET activation [218]. In addition, autocrine HGF/MET signaling in HCC promoted angiogenesis in cooperation with VEGF [219]. CRC and AML (acute myeloid leukemia) also showed autocrine MET upregulation accompanied by β-catenin and FGFR1 activation [220][221]. Downstream signaling pathways regulated by MET include the Ras/MAPK, Rac/JNK/p38, PI3K/AKT, STAT3, Src, and NF-κB pathways. Internalized MET either undergoes proteasomal degradation or continues to transmit signals in early endosomes and perinuclear compartments [222].
ATC cells (BRAFV600E mutant 8505C cell line) were resistant to vemurafenib treatment due to increased MET expression associated with reactivation of the PI3K/AKT pathway. Combination of vemurafenib with the MET inhibitor PHA665752 inhibited MET, p-AKT, and p-ERK, which led to improved in vitro anticancer activity against BRAF mutant ATC cells and sustained effects on an orthotopic BRAFV600E mutant ATC xenograft model [223]. BRAFV600E mutant ATC cells also showed EMT induction upon vemurafenib treatment by activation of AKT signaling (p-AKT increase) and upregulation of vimentin, β-catenin, and CD44, which was reversed by treatment with PHA665752 in combination with vemurafenib [224]. The recurrence of ATCs treated with the RAF/MEK inhibitor CH51222666766 depended on MET and HGF amplification, which was successfully treated with the MET inhibitors PF-04217903 and crizotinib. PF-04217903 showed in vivo activity against a MET-amplified ATC allograft model, while it remained inactive in MET-diploid allografts [225]. The phosphatase SHP2 upregulated MET expression in vemurafenib-resistant BRAFV600E thyroid cancers, which led to MAPK reactivation (Figure 2a) [47].
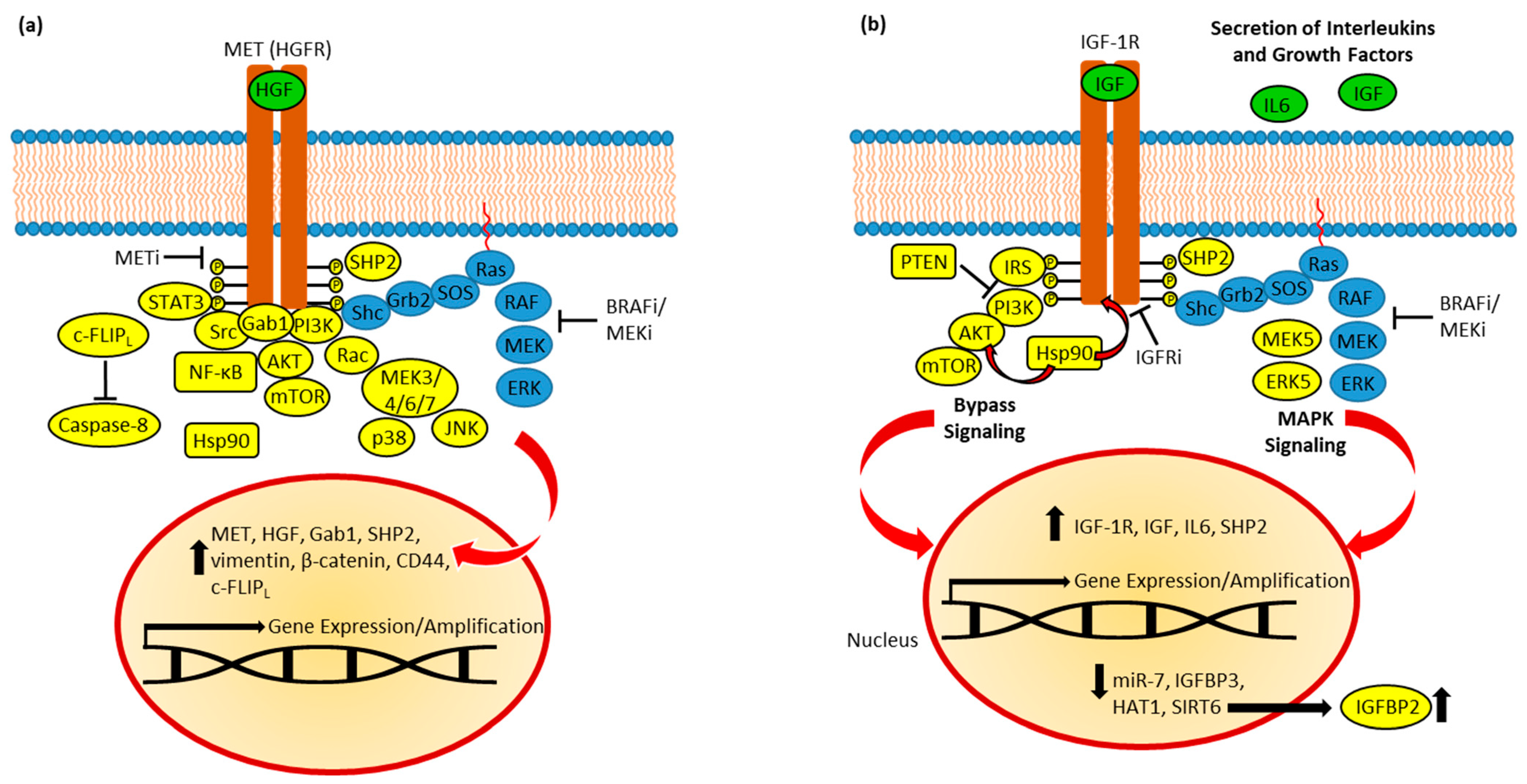
Figure 2. (a) MET in BRAFi resistance. Resistant cells established feedback loops and bypassed MAPK inhibition via AKT, STAT3, Rac-MEK, and Src signaling. Active SHP2 and Hsp90 promotes resistance, while c-FLIPL suppresses caspase-8-dependent apoptosis. Upregulated expression of HGF, MET, and Gab1 promote oncogenic signaling. EMT induction (upregulation of vimentin, β-catenin, and CD44) was observed. (b) IGF-1R in BRAFi resistance. Feedback leads to increased expression and secretion of IGFs and IL6, and upregulation of IGF-1R expression. SHP2 and induced MEK5/ERK5 signaling contribute to MAPK reactivation. PI3K/AKT signaling is upregulated and depends on IRS. Hsp90 stabilizes AKT and IGF-1R. Resistance was accompanied by suppression of miR-7, HAT1, and SIRT6; the latter caused upregulation of IGFBP2.
Primary resistance to vemurafenib in BRAFV600E mutant patient-derived melanoma cells was associated with upregulation of MET and Src signaling, as well as MET amplification [226]. The presence of secreted HGF in the tumor microenvironment led to MET-mediated MAPK and AKT reactivation, followed by RAF inhibitor resistance in BRAF mutant melanoma and various BRAF mutant CRC and glioblastoma cells [227]. In HGF-treated BRAFV600E melanoma cells, HGF was able to convey resistance to vemurafenib and combined dabrafenib plus trametinib treatment both in vitro and in vivo, while the addition of a MET inhibitor (dubbed compound 20 in that report) overcame HGF-mediated BRAF/MEK inhibitor resistance (Figure 2a). Notably, MET and Gab1 expression were increased upon treatment with vemurafenib and the MEK inhibitor PD0325901 in these BRAF mutant melanoma cells [228]. Since MET and ErbB3 are overexpressed and MET suppression led to EGFR activation in cutaneous malignant melanoma, crizotinib was studied in combination with the EGFR/HER2 inhibitor afatinib, which turned out to be a promising combination therapy in cutaneous melanoma independent from the BRAF mutation status. Afatinib plus crizotinib showed antiproliferative, pro-apoptotic, and anti-invasive activities in these melanoma cells, inhibited A375 tumor growth in vivo, and reduced WEE1 and IGF1R levels [229].
In BRAF mutant CRC, adaptive resistance to MEK inhibitors selumetinib and trametinib was correlated with increased MET/STAT3 signaling, followed by upregulation of c-FLIPL, which is an endogenous caspase-8 inhibitor. The anti-apoptotic effects of upregulated c-FLIPL were mediated by MET activation (increased p-MET) and were abrogated by a combination of selumetinib and the MET inhibitor crizotinib (Figure 2a). Since HDACs act as posttranscriptional c-FLIPL suppressors, the combination of selumetinib with the HDAC inhibitor entinostat led to increased BRAF mutant CRC cell death and to additive growth inhibition of BRAFV600E mutant HT-29 xenografts accompanied by caspase-3 activation [230].
Metastatic BRAFV600E mutant CRC patients treated with EGFR/BRAF inhibitor therapy developed resistance in line with MET amplification [231]. The combination of MET and BRAF inhibition led to a response in a BRAFV600E mutant MET-amplified CRC patient but developed acquired resistance after four months [232]. In the phase 1b/2 study of combined erlotinib plus verumafenib in BRAFV600E CRC patients, acquired resistance was observed in patients with MET amplification, among others [67]. Clinical studies revealed that MET-targeted therapies with selective MET inhibitors (e.g., capmatinib, tepotinib, and savolitinib) are especially promising in NSCLC with exon 14 mutation and in combination with EGFR inhibitors (e.g., osimertinib plus savolitinib or tepotinib plus geftinib) for the treatment of MET-mediated EGFR inhibitor-resistant NSCLC patients [233]. Yet, an EGFR-positive, MET-amplified NSCLC patient acquired the BRAF p.D594N mutation (a kinase-impaired mutation) by third-line osimertinib plus crizotinib therapy, which was successfully treated after replacing crizotinib with the MEK inhibitor trametinib. Although p.D594N impairs kinase activity, it activates MEK/ERK signaling, which could activate the downstream signaling of EGFR, followed by drug resistance [234]. Acquired MET inhibitor therapy resistance in MET exon 14-mutant NSCLC patients was based on activation of bypass signaling (including BRAF amplification) and on-target secondary mutations (kinase domain mutations and gene amplification) [235]. Combined vemurafenib plus crizotinib in (BRAF, MEK, and/or ERK inhibitor) pre-treated BRAF mutant advanced cancers was well tolerated and showed a response rate of 23% (melanoma and lung adenocarcinoma patients) [130].
6. IGFRs in BRAF Mutant Cancers
The insulin-like growth factor receptors (IGFRs) belong to the RTK family of insulin receptors. IGF-1R functions as a signal transducer following IGF-1 and IGF-2 ligand binding and is overexpressed in various cancers [236]. IGF-1R overexpression was observed in breast and prostate cancers, and its oncogenic activity was potentiated by estrogens and androgens. Ligand binding induces IGF-1R kinase activity, followed by autophosphorylation. Receptor substrates such as IRS (insulin receptor substrate) and Shc (Src-homology collagen) recognize certain receptor phospho-tyrosine residues and are phosphorylated by IGF-1R. Signal molecules with SH2 domains (PI3K, Grb2, and SHP2) bind to these IGF-1R substrate phospho-tyrosines and activate pathways such as MAPK and AKT signaling [237]. Autocrine loops were also described in certain cancers, e.g., acute myeloid leukemia, breast cancer, and endometrial carcinoma. IGF-1 and IGF-2 production by cancer cells activated IGF-1R, followed by increased proliferation and PI3K/AKT signaling [238][239][240]. In NSCLC, an autocrine loop based on IL-6 was identified, and IL-6 enhanced its own expression, followed by activation of IGF-1/2 and IGF-1R [241]. In short, IGF-1R is a key regulator of MAPK and PI3K/AKT signaling and plays a crucial role in resistance to MAPK inhibitors.
Prolonged exposure of BRAFV600E mutant melanoma to the BRAF inhibitor SB-590885 induced acquired resistance to SB-590885 and cross-resistance to another BRAF inhibitor (PLX4720). Persistent IGF-1R phosphorylation and activity were observed in resistant cells, while the negative regulator IGFBP3 was suppressed. Consequently, AKT signaling was activated by IGF-1R in the resistant melanoma cells, and the combination of a MEK inhibitor (GSK1120212 or selumetinib) with an IGF-1R inhibitor (cyclolignan picropodophyllin) suppressed the growth of BRAF inhibitor-resistant melanoma cells and spheroids [242]. Intrinsic and acquired resistance of BRAF mutant cutaneous melanoma cells to selumetinib was associated with AKT activation. Resistant cells showed high IGF-1R levels, and selumetinib-induced AKT upregulation was blocked by IGF-1R suppression [243]. BRAFV600E mutant A375 melanoma cells were initially sensitive to the ERK inhibitor SCH772984, but a resistant sub-line was developed by exposure to the inhibitor. ERK inhibitor resistance, which was accompanied by cross-resistance to vemurafenib and trametinib, was associated with upregulation of IGF-1R/MEK5/ERK5 signaling and activated IGF-1R, thus bypassing ERK1/2 suppression upon treatment with MAPK inhibitors. Notably, treatment of the ERK inhibitor-resistant melanoma with the IGF-1R inhibitor linsitinib suppressed ERK5 and tumor growth in vitro (3D spheroids) and in vivo, which offers a suitable therapy option for MAPK inhibitor-resistant melanoma (Figure 2b) [244]. Vice versa, linsitinib resistance in CRC was associated with strong activation of MAPK signaling, and the combination of the MEK1/2 inhibitor U0126 with linsitinib revealed synergistic effects in several CRC cell lines [245]. The Hsp90 inhibitor ganetespib suppressed the Hsp90 clients IGF-1R and AKT in melanoma cells, independent of the BRAF mutation status (Figure 2b). Ganetespib was also active against BRAF mutant cells with acquired BRAF-inhibitor resistance. It induced apoptosis and cell cycle arrest at the G1 and G2/M phases. Notably, ganetespib also suppressed EGFR, MET, BRAF, and CRAF in melanoma cells independent of the mutation status [246]. The bis-anthracycline WP760 exhibited high antiproliferative activity against a panel of melanoma cell lines, including vemurafenib-resistant cells, by suppressing IGF-1R and activating p53 [247]. Resistance of melanoma cells to treatment with the alkylating agent temozolomide was mediated by phosphorylated IGF-1R, and the IGF-1R inhibitors linsitinib and AZ3801 sensitized both BRAF mutant and wild-type melanoma cells to temozolomide, especially p53-wild-type cells. The combination of temozolomide with linsitinib led to distinct growth inhibition of BRAFV600E/p53-wild-type A375M melanoma xenografts [248]. PTEN loss-of-function mutation occurs in approximately 40% of BRAF mutant melanomas. Inhibition of IGF-1R in PTEN (LOF)/BRAF mutant melanoma augmented MAPK-targeted therapy efficacy in a synergistic way and limited glucose and insulin signaling [249]. In addition, IGF-1R inhibition combined with sorafenib exhibited over-additive antiproliferative effects on cholangiocarcinoma cells and appears to be a suitable strategy to improve sorafenib anticancer activity [250]. The phosphatase SHP2 upregulated IGF-1R in BRAFV600E thyroid cancers, accompanied by MAPK reactivation and vemurafenib resistance (Figure 2b) [47].
Absence of the histone acetyltransferase HAT1 conveyed BRAF inhibitor resistance in BRAFV600E mutant melanoma cells, which was mediated by increased IGF-1R activation followed by MAPK reactivation. Combination of the ERK inhibitor (SCH772984) with the IGF-1R inhibitor (BMS-754807) re-sensitized resistant BRAF mutant cells lacking the HAT1 gene to BRAF inhibitor treatment [251]. Sirtuins are NAD+-dependent histone deacetylases, and SIRT6 haploinsufficiency in BRAFV600E melanoma cells led to MAPK inhibitor resistance, which was mediated by upregulated IGF signaling. Reduced SIRT6 activity stabilized H3K56 acetylation in the IGFBP2 gene locus, leading to the promotion of IGFBP2 expression and IGF-1R activation (Figure 2b). Linsitinib plus dabrafinib suppressed IGF-1R and AKT phosphorylation and circumvented SIRT6 haploinsufficient melanoma resistance both in vitro and in vivo [252]. Vemurafenib resistance in BRAFV600E melanoma cells (A375) was associated with miR-7 suppression and upregulation of the miR-7 targets IGF-1R, EGFR, and CRAF, consequently leading to activation of MAPK and PI3K/AKT signaling [60].
Clinical trials with IGF-1R inhibitors (small molecules such as linsitinib and antibodies such as cixutumumab) have only shown modest results so far [253]. Notably, a phase 1 study with advanced solid tumors treated with cixutumumab in combination with selumetinib, which was well-tolerated by the participants, showed progression-free survival of more than 6 months in two of the three BRAF mutant cancer patients of the study, including a BRAF mutant thyroid cancer patient and a BRAF mutant CRC patient [254].
Table 4 summarizes the effects and mechanisms of the described FGFR, MET, and IGF-1R inhibitors.
Table 4. FGFR, MET and IGF-1R inhibitors and their activities in BRAF mutant cancers.
| Inhibitor | Mechanisms (Cancers/Cell Lines) | Clinical Studies | References |
|---|---|---|---|
| SU5402 and PD166866 (FGFR inhibitors) |
Antiproliferative and pro-apoptotic, suppressed p-AKT5 and p-STAT3, and synergy with sorafenib and BRAFV600E-selective inhibitor RG7204 (BRAFV600E melanomas) | - | [199] |
| Dovitinib (FGFR inhibitor) |
Suppression of p-FGFR1, p-ERK, and tumor growth (BRAFV600E mutant HT-29 xenograft), drug sensitization (vemurafenib-resistant RKO CRC cells), enforced vemurafenib and selumetinib activity (BRAF/MEK inhibitor-resistant BRAF mutant YUKSI melanoma cells, and vemurafenib-resistant YUMAC XR4MC8 melanoma xenograft) | - | [205][206] |
| BGJ-398 (FGFR inhibitor) |
Antiproliferative (BRAFV600E melanoma cells) | Currently being investigated (LOGIC-2 phase 2 trial with patients suffering from BRAF/MEK inhibitor-resistant advanced or metastatic BRAFV600E melanoma) | [203][210] |
| Aspirin | Suppression of FGFR, PI3K, and Notch signaling, reduced lesion size and number of distant colorectal cancer metastases (BrafV637E/+ and Villin-CreERT2/+ mice) | - | [207] |
| PHA665752 (MET inhibitor) |
Combination with vemurafenib inhibited MET, p-AKT, and p-ERK, increased anticancer activity (BRAF mutant ATC cells; orthotopic BRAFV600E mutant ATC xenograft), and combination with vemurafenib reversed EMT (BRAFV600E ATC cells) | - | [223][224] |
| Crizotinib (MET inhibitor) |
Active against recurrent ATCs treated with the RAF/MEK inhibitor, in combination with afatinib antiproliferative, pro-apoptotic, and anti-invasive activities (cutaneous melanoma cells), reduced WEE1 and IGF1R levels and suppressed in vivo tumor growth (A375 melanoma), and abrogation of anti-apoptotic c-FLIPL in combination with selumetinib (BRAF mutant CRC) | Well tolerated and response rate of 23% in combination with vemurafenib (BRAF/MEK inhibitor pre-treated BRAF mutant advanced melanoma and lung adenocarcinoma) | [130][225][229][230] |
| PF-04217903 (MET inhibitor) |
Active against recurrent ATCs treated with RAF/MEK inhibitor; in vivo activity against a MET-amplified ATC allograft model | - | [225] |
| Picropodophyllin (IGF-1R inhibitor) |
Combination with a MEK inhibitor (GSK1120212 or selumetinib) suppressed tumor growth (BRAF inhibitor-resistant melanoma cells and spheroids) | - | [242] |
| Linsitinib (IGF-1R inhibitor) |
Suppression of ERK5 and tumor growth in vitro (3D spheroids) and in vivo (MAPK inhibitor-resistant melanoma), synergy with MEK1/2 inhibitor U0126 (CRC cell lines), combination with AZ3801 sensitized cells to temozolomide (BRAF mutant and wild-type melanoma), combination with temozolomide led to distinct growth inhibition (BRAFV600E/p53-wild-type A375M melanoma xenografts), combination with dabrafinib suppressed p-IGF-1R and p-AKT, and breaks resistance in vitro and in vivo (SIRT6 haploinsufficient BRAFV600E melanoma) | Weak data so far | [244][245][248][252][253] |
| AZ3801 (IGF-1R inhibitor) |
Sensitization of BRAF mutant and BRAF wild-type (p53-wild-type) melanoma cells to temozolomide | - | [248] |
| Cixutumumab (IGF-1R inhibitor) |
- | Progression-free survival of more than 6 months in combination with selumetinib (phase 1, BRAF mutant thyroid cancer and CRC) | [254] |
7. Other RTKs and BRAF Mutant Cancers
7.1. AXL and MERTK
The AXL kinase (named after the Greek word for “uncontrolled”, anexelekto) is a TAM (TYRO3, AXL, and MERTK)-family RTK that is activated by the GAS6 (growth arrest-specific protein 6) ligand, resulting in receptor dimerization and autophosphorylation. Induced AXL activates PI3K/AKT, NF-κB, Ras/MAPK, Src/FAK, and JAK/STAT signaling, leading to cell proliferation and survival, cell migration, EMT, angiogenesis, drug resistance, cancer stem cell maintenance, and immune suppression [255]. AXL can also be activated by heterodimerization with ErbB, PDGFR, and MET and after phosphorylation by EGFR in GAS6-independent ways. In addition, post-transcriptional and post-translational regulation of AXL occurs by non-coding RNAs (miRNAs and lncRNAs), heat-shock proteins (Hsp90 and Hsp70), and proteolytic cleavage (by ADAM10 and ADAM17). High AXL expression was associated with drug resistance to ErbB inhibitors (osimertinib, erlotinib, cetuximab, and trastuzumab), checkpoint inhibitors (nivolumab), and chemotherapeutics (carboplatin and paclitaxel) in lung, breast, colorectal, endometrial, ovarian, and renal cancers. Thus, several AXL-targeted drugs and inhibitors, such as bemcentinib, merestinib, and sitravatinib, are currently in clinical trials [256].
In BRAF mutant melanoma, AXL/AKT signaling conveyed resistance to vemurafenib in PTEN-wild-type cells but not in PTEN-impaired cells. Activating phosphorylation of AXL was observed. The selective AXL inhibitor bemcentinib (R428 and BGB324) suppressed AXL and AKT phosphorylation and re-sensitized resistant melanoma cells and tumors to vemurafenib both in vitro and in vivo [257]. High expression of AXL was discovered in BRAF mutant melanoma lymph node metastases, and the anticancer activity of vemurafenib against the resistant melanoma models was augmented by combination with bemcentinib in vitro, ex vivo, and in vivo, which was associated with suppressed phospho-ERK and phospho-S6 (an mTORC1 downstream substrate) levels. Bemcentinib enhanced vemurafenib-based apoptosis induction and increased ferroptosis, while suppressing autophagy [258]. Upregulation of NF-κB was responsible for the increased expression of AXL in BRAF inhibitor-resistant BRAFV600 mutant melanoma, which was successfully treated with AXL inhibitors such as bemcentinib, cabozantinib, and XL880, accompanied by suppression of p-AKT and p-ERK levels [259]. The anti-AXL antibody drug conjugate with the microtubule-disrupting agent MMAE (monomethyl auristatin E), AXL-107-MMAE, displayed cooperative activity in combination with BRAF/MEK inhibitors against melanoma. In particular, BRAF/MEK inhibitors upregulated AXL transcription, thus sensitizing melanoma to AXL-107-MMAE treatment [260].
In addition to AXL, MERTK (Mer tyrosine kinase), another TAM family kinase and a main regulator of phagocytosis/efferocytosis, was upregulated in acquired BRAF/MEK inhibitor-resistant melanoma. BRAF inhibition activated the EMT regulator Zeb2 in BRAFV600E mutant melanoma, which upregulated MERTK via mTORC1-induced autophagy. Consequently, the combination of PLX4720 with the autophagy inhibitor chloroquine led to increased tumor growth inhibition in BRAFV600E melanoma xenografts [261]. In BRAF mutant melanoma cells, the MERTK inhibitor UNC2025 in combination with vemurafenib induced cell death and suppressed efficiently ERK, AKT, and STAT6. In addition, UNC2025 plus vemurafenib inhibited BRAF mutant melanoma xenograft growth and was well tolerated by mice. Since UNC2025 was active independent of the BRAF mutation status, it can become a therapy option where BRAF inhibitor therapy fails [262].
AXL has a large homology to RET, and RET mutants are relevant for thyroid cancer and NSCLC progression. The phosphatase SHP2 upregulated AXL and RET in vemurafenib-resistant BRAFV600E thyroid cancers [47]. Acquired resistance to selpercatinib (RET inhibitor) treatment was associated with MET amplification in RET mutant cancers; however, selpercatinib plus a MET inhibitor (crizotinib) led to a BRAFD594N mutant subclone. The D594N mutant is a kinase-dead RAS- and BRAF dimer-dependent BRAF mutant that re-establishes the Ras/MAPK dependence of RET-driven tumors [263].
7.2. ALK
Anaplastic lymphoma kinase (ALK) is another important RTK mediator of BRAF inhibitor resistance. Gene fusions (e.g., EML4-ALK) and amplifications, as well as activating mutations (e.g., F1174L/V and F1245C/L/V), are the most abundant ALK genetic alterations found in various cancers such as neuroblastomas, lymphomas, lung cancer, or renal cell carcinoma [264][265][266][267]. Four ALK inhibitors—crizotinib, ceritinib, alectinib, and brigatinib—were approved for the therapy of ALK-positive NSCLC [268].
It was shown that acquired vemurafenib resistance in BRAF mutant melanoma was mediated by ALK/PI3K/AKT signaling since activated p-ALK and downstream p-AKT levels were increased and the ALK ligand FAM150A was required. Ceritinib suppressed vemurafenib-resistant melanoma in vitro and in vivo. Yet, the formation of dual vemurafenib-ceritinib resistance based on suppressed apoptosis (upregulated BCL2, cIAP1/2, and survivin) was observed, which could be treated with the anti-apoptotic protein inhibitor AT101 [269]. In addition, a patient with EML4-ALK fusion and BRAFV600E mutant NSCLC, whose disease progressed upon first-line treatment with dabrafenib plus trametinib, responded well to alectenib (complete remission, progression-free survival for over 26 months) [270]. Vice versa, ALK-positive NSCLC patients receiving ALK-targeted therapy developed BRAF mutations. Cricotinib was administered to a patient with EML4-ALK-rearranged lung adenocarcinoma for 20 months before the tumor progressed. Because alectinib therapy also resulted in early progression and lorlatinib was inefficient, analysis of the resistant tumor revealed new activating BRAFV600E and ALKI1171T mutations, which were associated with the acquired ALK inhibitor resistance [271]. Another lung adenocarcinoma patient with an EML4-AXL fusion tumor developed acquired AXL inhibitor resistance upon second-line crizotinib and third-line alectinib treatment by a rare A598-T599insV mutation of BRAF. A fourth-line treatment with a combined BRAF/MEK inhibitor was also unsuccessful, probably because of the EML4-AXL fusion, which causes BRAF/MEK inhibitor resistance [272].
7.3. DDR
Discoidin domain receptors (DDRs, not to be mixed with DDR/DNA damage response) are unique RTKs because they are activated by collagen binding. DDRs are major sensors of cell–collagen interactions and the regulation of cell traffic in tissue compartments, and, thus, DDR signaling affects cancer progression in terms of cell proliferation, migration, invasion, and drug resistance. There are two forms of DDR: DDR1 (five isoforms) and DDR2 (one isoform). DDRs consist of an extracellular discoidin domain, a transmembrane domain, and an intracellular kinase domain. Upon collagen binding, DDRs dimerize and undergo activating autophosphorylation. DDRs can also transmit signals independent of functional kinases. DDR1 activation upregulates the PI3K/AKT and Ras/MAPK pathways, while DDR2 regulates Src signaling. Point mutations and gene amplifications contribute to the deregulation of DDR signaling in various cancers [273].
The pan-DDR inhibitor DDR1-IN-1 was able to suppress BRAF mutant melanoma xenograft growth [274]. DDR1 expression was higher in BRAF mutant melanomas, and DDRs appear to be suitable anticancer drug targets in melanomas, which have a collagen-rich stroma [275]. Matrix-mediated vemurafenib and trametinib resistance in BRAFV600E melanoma was correlated with DDR1/2 activation and phosphorylation. The DDR-dependent drug resistance was associated with pro-survival NIK/IKKα/NF-κB2 activation. Treatment of BRAFV600E melanoma with the DDR-inhibitory multikinase inhibitor imatinib broke the BRAF/MEK inhibitor resistance induced by the extracellular matrix. Imatinib also augmented vemurafenib anticancer activity against BRAFV600E melanoma xenografts by suppressing collagen remodeling [276]. It was also shown that the acquired resistance of NRAS mutant melanomas to trametinib was mediated by DDR1 activation [277]. DDR1 and DDR2 were overexpressed together with ephedrine receptors in double vemurafenib- and MEK inhibitor-cobimetinib-resistant BRAFV600E melanoma. The multikinase inhibitor ALW-II-41-27 showed antiproliferative and anti-migratory activities and suppressed DDR1/2 phosphorylation as well as phospho-AKT and phospho-MEK levels in the drug-resistant cells [278].
7.4. EphA2
Last but not least, ephrin receptors such as EphA2 are RTKs, which attracted increased interest as drivers of BRAF inhibitor resistance. Eph receptors bind the membrane-located ephrin ligand of an adjacent cell and regulate cell–cell interactions. In particular, the EphA2 gene and protein are overexpressed in several cancer types and play a crucial role in carcinogenesis. Several treatment options for EphA2-targeting, including kinase inhibitors and various biologics, are currently in clinical trials [279]. EphA2 prefers interaction with ephrin A1, and canonical ephrin A1-dependent activation of EphA2 leads to dimerization and transphosphorylation but is tumor-suppressive. However, EphA2 can also be activated by other signaling molecules (e.g., Wnt ligands) and upregulate Ras/MAPK and AKT signaling [280]. Non-canonical ligand-independent effector roles were described for EphA2 downstream from MEK/ERK and upon S897 phosphorylation by AKT [281][282].
Vemurafenib-resistant BRAFV600E melanomas depended on EphA2, which was upregulated and phosphorylated in cells with acquired resistance, leading to enhanced tumor cell migration. The EphA2 inhibitor ALW-II-41-27 inhibited vemurafenib-resistant tumor cell growth and showed synergistic effects in combination with vemurafenib, while resistant cells pre-treated with ALW-II-41-27 were re-sensitized to vemurafenib treatment. EphA2 inhibition suppressed phospho-AKT and phospho-ERK levels and had pro-apoptotic effects. Moreover, ALW-II-41-27 inhibited the growth of BRAFV600E mutant vemurafenib-resistant and -sensitive melanoma xenografts [283]. Vemurafenib-resistant BRAFV600E melanomas induced EphA2 by S897 phosphorylation through AKT, which strongly fostered metastasis formation. In addition, high EphA2 levels were found in BRAFV600E melanoma patient metastases upon BRAF inhibitor treatment [284]. Interestingly, proteomic analyses of dabrafenib-resistant BRAFV600E melanoma cells showed downregulated EphA2 expression when compared with sensitive cells, probably in order to prevent canonical EphA2 tumor suppressor signaling associated with MAPK downregulation [285]. Non-canonical EphA2 signaling in BRAF inhibitor-resistant melanoma cells was driven by AKT and HDAC8, which led to upregulation of EphA2 and Cdc42 and revealed increased endothelial cell adhesion and transendothelial migration associated with an amoeboid cell phenotype enabling metastasis formation [286]. Acquired BRAF inhibitor-resistant BRAF mutant melanoma cells exhibited increased expression of ETS transcription factor FLI1 and aminopeptidase-N (CD13/ANPEP) in addition to upregulated EphA2 and overexpressed MET. Inhibition of CD13/ANPEP with an antibody downregulated phospho-EphA2, as well as phospho-AKT and phospho-RSK, leading to apoptosis induction and reduced tumor cell migration. FLI1 silencing also reduced phospho-EphA2 and re-sensitized resistant cells to vemurafenib [287]. EphA2 was overexpressed together with DDR1 and DDR2 in double vemurafenib- and cobimetinib (MEK inhibitor)-resistant BRAFV600E melanoma, and treatment of the drug-resistant cells with ALW-II-41-27 downregulated phospho-EphA2 together with phospho-AKT and phospho-MEK levels [278].
The multikinase inhibitor dasatinib was also identified as an EphA2 inhibitor. Because most of dasatinib’s anticancer properties were attributed to its Src family and BCR-ABL inhibitory activities, some of its anti-metastatic properties, e.g., in BRAF inhibitor-resistant melanomas, might be correlated with EphA2-targeting [51]. Dasatinib resistance in uterine cancers was associated with increased EphA2 expression and MAPK activation, which was overcome by MEK inhibition (trametinib) [288]. It was shown that the crosstalk between BRAF and EphA2 is crucial for dasatinib sensitivity in uterine cancer cells, and sensitivity was accompanied by increased levels of caveolin-1 (CAV-1), which suppresses BRAF/CRAF dimer levels and MAPK signaling [289].
A BRAF mutant melanoma patient with brain metastases developed resistance to first-line treatment with dabrafinib and trametinib associated with upregulated EphA2 expression and increased phospho-EphA2 levels, which was reversible and sensitive to rechallenge treatment with encorafenib plus binimetinib [19]. Yet, EphA2-targeted clinical data is still rare. An early phase 1 study with the antibody conjugate MEDI-547 (an anti-EphA2 antibody linked with the toxin auristatin) in advanced or refractory solid tumors was terminated due to toxicity [290]. In addition, a phase 1 study of the anti-EphA2 antibody DS-8895a in patients with advanced EphA2-positive cancers showed only limited activity [291]. The clinically approved anti-leukemia agent dasatinib appears to be a promising drug in terms of EphA2-targeting in BRAF mutant cancers. Notably, an NSCLC patient with a kinase impaired (non-V600) BRAFY472C mutation responded enormously well to dasatinib therapy and remained cancer-free for at least four years [292]. In addition, a phase 2 trial of dasatinib combined with dendritic cell vaccines against TBVA (tumor blood vessel antigen), which targets EphA2, among others, was well-tolerated and revealed an immune response (CD8+ T cell response) together with a clinical response in 46% of the studied checkpoint-refractory advanced melanoma patients [293]. This study included BRAF mutant cancer patients, and, thus, EphA2-targeting dasatinib might also be taken into account for the therapy of other BRAF mutant cancers in the future.
Table 5 summarizes the effects and mechanisms of the described AXL, AKT, DDR, and EphA2 inhibitors.
Table 5. AXL, ALK, DDR, and EphA2 inhibitors and their activities in BRAF mutant cancers.
| Inhibitor | Mechanisms (Cancers/Cell Lines) | Clinical Studies | References |
|---|---|---|---|
| Bemcentinib (AXL inhibitor) |
Suppression of p-AXL and p-AKT; re-sensitization to vemurafenib (resistant melanoma cells and tumors); suppressed p-ERK and p-S6, augmented vemurafenib activity (in vitro, ex vivo, and in vivo); enhanced apoptosis and ferroptosis; suppressed autophagy (resistant melanoma); suppression of p-AKT and p-ERK (BRAF inhibitor-resistant BRAFV600 mutant melanoma) | Clinical trials started and ongoing | [256][257][258][259] |
| Cabozantinib (AXL inhibitor) |
Suppression of p-AKT and p-ERK (BRAF inhibitor-resistant BRAFV600 mutant melanoma) | - | [259] |
| XL880 (AXL inhibitor) |
Suppression of p-AKT and p-ERK (BRAF inhibitor-resistant BRAFV600 mutant melanoma) | - | [259] |
| AXL-107-MMAE (AXL inhibitor) |
Cooperative activity in combination with AXL-upregulating BRAF/MEK inhibitors (melanoma) | - | [260] |
| UNC2025 (AXL inhibitor) |
Induction of cell death; suppression of ERK, AKT, STAT6, and tumor growth in combination with vemurafenib (BRAF mutant melanoma cells and xenografts) | - | [262] |
| Ceritinib (ALK inhibitor) |
Suppression of vemurafenib resistance in vitro and in vivo; formation of dual vemurafenib–ceritinib resistance by suppressed apoptosis overcome by pro-apoptotic AT101 (melanoma) | - | [262] |
| Alectenib (ALK inhibitor) |
- | Complete remission and progression-free survival for over 26 months (case report, pretreated EML4-ALK fusion and BRAFV600E mutant NSCLC) | [270] |
| DDR1-IN-1 (DDR inhibitor) |
Suppression of BRAF mutant melanoma xenografts | - | [274] |
| Imatinib (DDR inhibitor) |
Breach of BRAF/MEK inhibitor resistance induced by the extracellular matrix (BRAFV600E melanoma); increased vemurafenib activity by suppression of collagen remodeling (BRAFV600E melanoma xenografts) | - | [276] |
| ALW-II-41-27 (DDR inhibitor) |
Antiproliferative and anti-migratory activities; suppression of p-DDR1/2, p-AKT, and p-MEK (vemurafenib/cobimetinib-resistant BRAFV600E melanoma) | - | [278] |
| ALW-II-41-27 (EphA2 inhibitor) |
Synergy; re-sensitization to vemurafenib; suppression of p-AKT and p-ERK, pro-apoptotic effects; tumor growth inhibition (BRAFV600E mutant vemurafenib-resistant and -sensitive melanoma cells and xenografts); downregulation of p-EphA2, p-AKT, and p-MEK (vemurafenib/cobimetinib-resistant BRAFV600E melanoma) | - | [278][283] |
| Dasatinib (EphA2 inhibitor) |
Sensitivity accompanied by MAPK signaling suppressor caveolin-1 upregulation (uterine cancer cells) | High response and disease-free for a min. of 4 years (case report, patient with kinase impaired non-V600 BRAFY472C mutation NSCLC); immune response (CD8+ T cell response) and clinical response (46%) in combination with dendritic cell vaccines (phase 2 trial, advanced checkpoint-refractory and BRAF mutant melanoma) | [289][292][293] |
References
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26.
- Kumar, V.; Abbas, A.; Aster, J. Robbins Basic Pathology; Elsevier/Saunders: Philadelphia, PA, USA, 2013; p. 179.
- Wittinghofer, A.; Waldmann, H. Ras—A molecular switch in tumor formation. Angew. Chem. Int. Ed. 2000, 39, 4192–4214.
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 2001, 8, 11–31.
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56.
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2014, 4, 36–54.
- Lin, Y.; Wang, X.; Jin, H. EGFR-TKI resistance in NSCLC patients: Mechanisms and strategies. Am. J. Cancer Res. 2014, 4, 411–435.
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401.
- Johnson, G.L.; Stuhlmiller, T.J.; Angus, S.P.; Zawistowski, J.S.; Graves, L.M. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 2014, 20, 2516–2522.
- Kim, S.; Carvajal, R.; Kim, M.; Yang, H.W. Kinetics of RTK activation determine ERK reactivation and resistance to dual BRAF/MEK inhibition in melanoma. Cell Rep. 2023, 42, 112570.
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235.
- Held, M.A.; Langdon, C.G.; Platt, J.T.; Graham-Steed, T.; Liu, Z.; Chakraborty, A.; Bacchiocchi, A.; Koo, A.; Haskins, J.W.; Bosenberg, M.W.; et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 2013, 3, 52–67.
- Kennessey, I.; Kramer, Z.; István, L.; Cserepes, M.T.; Garay, T.; Hegedüs, B.; Dobos, J.; Tímár, J.; Tóvári, J. Inhibition of epidermal growth factor receptor improves antitumor efficacy of vemurafenib in BRAF-mutant human melanoma in preclinical model. Melanoma Res. 2018, 28, 536–546.
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.J.M.; et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF mutant colorectal cancer. Cancer Res. 2016, 76, 4504–4515.
- Tiedt, R.; King, F.J.; Stamm, C.; Niederst, M.J.; Delach, S.; Zumstein-Mecker, S.; Meltzer, J.; Mulford, I.J.; Labrot, E.; Schacher Engstler, B.; et al. Integrated CRISPR screening and drug profiling identifies combination opportunities for EGFR, ALK, and BRAF/MEK inhibitors. Cell Rep. 2023, 42, 112297.
- Molnár, E.; Garay, T.; Donia, M.; Baranyi, M.; Rittler, D.; Berger, W.; Timár, J.; Grusch, M.; Hegedüs, B. Long-term vemurafenib exposure induced alterations of cell phenotypes in melanoma: Increased cell migration and its association with EGFR expression. Int. J. Mol. Sci. 2019, 20, 4484.
- Li, B.; Jin, J.; Guo, D.; Tao, Z.; Hu, X. Immune checkpoint inhibitors combined with targeted therapy: The recent advances and future potentials. Cancers 2023, 15, 2858.
- Dugo, M.; Nicolini, G.; Tragni, G.; Bersani, I.; Tomassetti, A.; Colonna, V.; Del Vecchio, M.; De Braud, F.; Canevari, S.; Anichini, A.; et al. A melanoma subtype with intrinsic resistance to BRAF inhibition identified by receptor tyrosine kinase gene-driven classification. Oncotarget 2015, 6, 5118–5133.
- Fukushima, H.; Iwata, Y.; Saito, K.; Sugiura, K. Successful rechallenge therapy for BRAF/MEK inhibitor resistant multiple brain metastases of melanoma. J. Dermatol. 2021, 48, 1291–1295.
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with Boolean in silico models. Cancers 2020, 12, 28.
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541.
- Song, Q.; Sun, X.; Guo, H.; Yu, Q. Concomitant inhibition of receptor tyrosine kinases and downstream AKT synergistically inhibited growth of KRAS/BRAF mutant colorectal cancer cells. Oncotarget 2017, 8, 5003–5015.
- Cheng, L.; Jin, Y.; Liu, M.; Ruan, M.; Chen, L. HER inhibitor promotes BRAF/MEK inhibitor-induced redifferentiation in papillary thyroid cancer harboring BRAFV600E. Oncotarget 2017, 8, 19843–19854.
- Miele, E.; Abballe, L.; Spinelli, G.P.; Besharat, Z.M.; Catanzaro, G.; Chiacchiarini, M.; Vacca, A.; Po, A.; Capalbo, C.; Ferretti, E. BRAF mutant colorectal cancer: ErbB2 expression levels as predictive factor for the response to combined BRAF/ErbB inhibitors. BMC Cancer 2020, 20, 129.
- Dent, P.; Booth, L.; Poklepovic, A.; Kirkwood, J.M. Neratinib kills B-RAF V600E melanoma via ROS-dependent autophagosome formation and death receptor signaling. Pigment Cell Melanoma Res. 2022, 35, 66–77.
- Ng, Y.-K.; Lee, J.-Y.; Supko, K.M.; Khan, A.; Torres, S.M.; Berwick, M.; Ho, J.; Kirkwood, J.M.; Siegfried, J.M.; Stabile, L.P. Pan-erbB inhibition potentiates BRAF inhibitors for melanoma treatment. Melanoma Res. 2014, 24, 207–218.
- Cronise, K.E.; Hernandez, B.G.; Gustafson, D.L.; Duval, D.L. Identifying the ErbB/MAPK signaling cascade as a therapeutic target in canine bladder cancer. Mol. Pharmacol. 2019, 96, 36–46.
- Okimoto, R.A.; Lin, L.; Olivas, V.; Chan, E.; Markegard, E.; Rymar, A.; Neel, D.; Chen, X.; Hemmati, G.; Bollag, G.; et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 13456–13461.
- Tang, Z.; Yuan, X.; Du, R.; Cheung, S.-H.; Zhang, G.; Wei, J.; Zhao, Y.; Feng, Y.; Peng, Y.; Zhang, Y.; et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol. Cancer Ther. 2015, 14, 2187–2197.
- Kotani, H.; Adachi, Y.; Kitai, H.; Tomida, S.; Bando, H.; Faber, A.C.; Yoshino, T.; Voon, D.C.; Yano, S.; Ebi, H. Distinct dependencies on receptor tyrosine kinases in the regulation of MAPK signaling between BRAF V600E and non-V600E mutant lung cancers. Oncogene 2018, 37, 1775–1787.
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J. Clin. Oncol. 2020, 38, 2140–2150.
- Yuan, X.; Tang, Z.; Du, R.; Yao, Z.; Cheung, S.-H.; Zhang, X.; Wie, J.; Zhao, Y.; Du, Y.; Liu, Y.; et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in KRAS mutant tumors. Mol. Oncol. 2020, 14, 1833–1849.
- Aboubakar Nana, F.; Ocak, S. Targeting BRAF activation as acquired resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR-mutant non-small-cell lung cancer. Pharmaceutics 2021, 13, 1478.
- Ribeiro, M.F.S.A.; Knebel, F.H.; Bettoni, F.; Saddi, R.; Sacardo, K.P.; Canedo, F.S.N.A.; Alessi, J.V.M.; Shimada, A.K.; Marin, J.F.G.; Camargo, A.A.; et al. Impressive response to dabrafenib, trametinib, and osimertinib in a metastatic EGFT-mutant/BRAF V600E lung adenocarcinoma patient. NPJ Precis. Oncol. 2021, 5, 5.
- Zeng, R.; Luo, L.; Sun, X.; Bao, Z.; Du, W.; Dai, R.; Tang, W.; Gao, B.; Xiang, Y. EGFR/BRAF/MEK co-inhibition for EGFR-mutated lung adenocarcinoma patients with an acquired BRAFV600E mutation: A case report and review of literature. Cancer Drug Resist. 2021, 4, 1019–1027.
- Orciulo, C.; Cappuzzo, F.; Landi, L.; Resuli, B.; Carpano, S.; Vidiri, A.; Buglioni, S.; Mandoj, C.; Ciliberto, G.; Minuti, G. Pretreated EGFRdel19/BRAFV600E lung adenocarcinoma with leptomeningeal disease achieving long-lasting disease control on osimertinib, dabrafenib, and trametinib: A case report. JTO Clin. Res. Rep. 2023, 4, 100545.
- Leduc, C.; Merlio, J.P.; Besse, B.; Blons, H.; Debieuvre, D.; Bringuier, P.P.; Monnet, I.; Rouquette, I.; Fraboulet-Moreau, S.; Lemoine, A.; et al. Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: Results of the nationwide French Cooperative Intergroup (IFCT) program. Ann. Oncol. 2017, 28, 2715–2724.
- Kong, W.-M.; Guo, Y.-J.; Ma, J.; Shi, C. BTN2A1-BRAF fusion may be a novel mechanism of resistance to osimertinib in lung adenocarcinoma: A case report. Transl. Cancer Res. 2023, 12, 186–193.
- Kirkpatrick, P.; Graham, J.; Muhsin, M. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550.
- Kong, L.; Zhang, Q.; Mao, J.; Cheng, L.; Shi, X.; Yu, L.; Hu, J.; Yang, M.; Li, L.; Liu, B.; et al. A dual-targeted molecular therapy of PP242 and cetuximab plays an anti-tumor effect through EGFR downstream signaling pathways in colorectal cancer. J. Gastrointest. Oncol. 2021, 12, 1625–1642.
- Veluchamy, J.P.; Spanholtz, J.; Tordoir, M.; Thijssen, V.L.; Heideman, D.A.M.; Verheul, H.M.W.; de Gruijl, T.D.; van der Vliet, H.J. Combination of NK cells and cetuximab to enhance anti-tumor responses in RAS mutant metastatic colorectal cancer. PLoS ONE 2016, 11, e0157830.
- Wu, Z.; Huang, M.; Gong, Y.; Lin, C.; Guo, W. BRAF and EGFR inhibitors synergize to increase cytotoxic effects and decrease stem cell capacities in BRAF(V600E)-mutant colorectal cancer cells. Acta Biochim. Biophys. Sin. 2018, 50, 355–361.
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238.
- Chung, Y.-C.; Chiu, H.-H.; Wei, W.-C.; Chang, K.-J.; Chao, W.-T. Application of trastuzumab emtansine in HER-2-positive and KRAS/BRAF-mutated colon cancer cells. Eur. J. Clin. Investig. 2020, 29, e13255.
- Tchekmedyian, V.; Dunn, L.; Sherman, E.; Baxi, S.S.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Enhancing radioiodine incorporation in BRAF-mutant, radioiodine-refractory thyroid cancers with vemurafenib and the anti-ErbB3 monoclonal antibody CDX-3379: Results of a pilot clinical trial. Thyroid 2022, 32, 273–282.
- Roller, D.G.; Capaldo, B.; Bekiranov, S.; Mackey, A.J.; Conaway, M.R.; Petricoin, E.F.; Gioeli, D.; Weber, M.J. Combinatorial drug screening and molecular profiling reveal diverse mechanisms of intrinsic and adaptive resistance to BRAF inhibition in V600E BRAF mutant melanomas. Oncotarget 2015, 7, 2734–2753.
- Ma, W.; Tian, M.; Hu, L.; Ruan, X.; Zhang, W.; Zheng, X.; Gao, M. Early combined SHP2 targeting reverses the therapeutic resistance of vemurafenib in thyroid cancer. J. Cancer 2023, 14, 1592–1604.
- Invrea, F.; Punzi, S.; Petti, C.; Minelli, R.; Peoples, M.D.; Bristow, C.A.; Vurchio, V.; Corrado, A.; Bragoni, A.; Marchiò, C.; et al. Synthetic lethality screening highlights colorectal cancer vulnerability to concomitant blockade of NEDD8 and EGFR pathways. Cancers 2021, 13, 3805.
- Forsythe, N.; Refaat, A.; Javadi, A.; Khawaja, H.; Weir, J.-A.; Emam, H.; Allen, W.L.; Burkamp, F.; Popovici, V.; Jithesh, P.V.; et al. The unfolded protein response: A novel therapeutic target for poor prognostic BRAF mutant colorectal cancer. Mol. Cancer Ther. 2018, 17, 1280–1290.
- Delgado-Goni, T.; Galobart, T.C.; Wantuch, S.; Normantaite, D.; Leach, M.O.; Whittaker, S.R.; Beloueche-Babari, M. Increased inflammatory lipid metabolism and anaplerotic mitochondrial activation follow acquired resistance to vemurafenib in BRAF-mutant melanoma cells. Br. J. Cancer 2020, 122, 72–81.
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajilic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167.
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256.
- Garcia-Rendueles, M.E.R.; Krishnamoorthy, G.; Saqcena, M.; Acuna-Ruiz, A.; Revilla, G.; de Stanchina, E.; Knauf, J.A.; Lester, R.; Xu, B.; Ghossein, R.A.; et al. Yap governs a lineage-specific neuregulin1 pathway-driven adaptive resistance to RAF kinase inhibitors. Mol. Cancer 2022, 21, 213.
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cells 2014, 158, 171–184.
- Chen, H.; Chen, W.; Zhang, X.; Hu, L.; Tang, G.; Kong, J.; Wang, Z. E26 transformation (ETS)-specific related transcription factor-3 (ELF3) orchestrates a positive feedback loop that constitutively activates the MAPK/Erk pathway to drive thyroid cancer. Oncol. Rep. 2019, 41, 570–578.
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122.
- Abel, E.V.; Basile, K.J.; Kugel III, C.H.; Witkiewicz, A.K.; Le, K.; Amaravadi, R.K.; Karakousis, G.C.; Xu, X.; Lu, W.; Schuchter, L.M.; et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Investig. 2013, 123, 2155–2168.
- Herr, R.; Halbach, S.; Heizmann, M.; Busch, H.; Boerries, M.; Brummer, T. BRAF inhibition upregulates a variety of receptor tyrosine kinases and their downstream effector Gab2 in colorectal cancer cell lines. Oncogene 2018, 37, 1576–1593.
- Dang, H.; Sui, M.; He, Q.; Xie, J.; Liu, Y.; Hou, P.; Ji, M. Pin1 inhibitor API-1 sensitizes BRAF-mutant thyroid cancers to BRAF inhibitors by attenuating HER3-mediated feedback activation of MAPK/ERK and PI3K/AKT pathways. Int. J. Biol. Macromol. 2023, 248, 125867.
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget 2016, 7, 53558–53570.
- Liu, S.; Tetzlaff, M.T.; Wang, T.; Yang, R.; Xie, L.; Zhang, G.; Krepler, C.; Xiao, M.; Beqiri, M.; Xu, W.; et al. miR-200c/Bmi1 axis and epithelial-mesenchymal transition, contribute to acquired resistance to BRAF inhibitor treatment. Pigment Cell Melanoma Res. 2015, 28, 431–441.
- Biersack, B. Interactions between anticancer active platinum complexes and non-coding RNAs/microRNAs. Non-Coding RNA Res. 2017, 2, 1–17.
- Biersack, B. Alkylating anticancer agents and their relations to microRNAs. Cancer Drug Resist. 2019, 2, 1–17.
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657.
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: Updated survival results and subgroup analyses from the BEACON study. J. Clin. Oncol. 2021, 39, 273–284.
- Cho, S.M.; Esmail, A.; Abdelrahim, M. Triple-regimen of verumafenib, irinotecan, and cetuximab for the treatment of BRAFV600E-mutant CRC: A case report and review. Front. Pharmacol. 2021, 12, 795381.
- Tan, L.; Tran, B.; Tie, J.; Markman, B.; Ananda, S.; Tebbutt, N.C.; Michael, M.; Link, E.; Wong, S.Q.; Chandrashekar, S.; et al. A phase Ib/II trial of combined BRAF and EGFR inhibition in BRAF V600E positive metastatic colorectal cancer and other cancers: The EVICT (erlotinib and vemurafenib in combination trial) study. Clin. Cancer Res. 2023, 29, 1017–1030.
- Martini, G.; Ciardiello, D.; Napolitano, S.; Martinelli, E.; Troiani, T.; Latiano, T.P.; Acallone, A.; Normanno, N.; Di Maio, M.; Maiello, E.; et al. Efficacy and safety of a biomarker-driven cetuximab-based treatment regimen over 3 treatment lines in mCRC patients with RAS/BRAF wild type tumors at start of first line: The CAPRI 2 GOIM trial. Front. Oncol. 2023, 13, 1069370.
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.-J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; MAnyam, G.; Hong, D.S.; et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294.
- Xu, Y.; Wang, G.; Zheng, X.; Chang, W.; Fu, J.; Zhang, T.; Lin, Q.; Lv, Y.; Zhu, Z.; Tang, W.; et al. Treatment of metastatic colorectal cancer with BRAF V600E mutation: A multicenter real-world study in China. Eur. J. Surg. Oncol. 2023, 49, 106981.
- Grassilli, E.; Cerrito, M.G. Emerging actionable targets to treat therapy-resistant colorectal cancers. Cancer Drug Resist. 2022, 5, 36–63.
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.; Bando, H.; Van Seventer, E.; Taniguchi, H.; Zhao, H.Y.; Thant, C.; de Stanchina, E.; et al. Response to anti-EGFR therapy in patients with BRAF non-V600 mutant metastatic colorectal cancer. Clin. Cancer Res. 2019, 25, 7089–7097.
- Wang, Y.; Jones, J.C.; Kipp, B.R.; Grothey, A. Activity of EGFR antibody in non-V600 BRAF mutant metastatic colorectal cancer. Ann. Oncol. 2019, 30, 148–149.
- Randon, G.; Intini, R.; Cremolini, C.; Elez, E.; Overman, M.J.; Lee, J.; Manca, P.; Bergamo, F.; Pagani, F.; Antista, M.; et al. Tumour mutational burden predicts resistance to EGFR/BRAF blockade in BRAF-mutated microsatellite stable metastatic colorectal cancer. Eur. J. Cancer 2022, 161, 90–98.
- Dankner, M.; Wang, Y.; Fazelzad, R.; Johnson, B.; Nebhan, C.A.; Dagogo-Jack, I.; Myall, N.J.; Richtig, G.; Bracht, J.W.P.; Gerlinger, M.; et al. Clinical activity of mitogen.activated protein kinase-targeted therapies in patients with non-V600 BRAF-mutant tumors. JCO Precis. Oncol. 2022, 6, e2200107.
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473.
- Farhan, M. Insights on the role of polyphenols in combating cancer drug resistance. Biomedicines 2023, 11, 1709.
- Lev-Ari, S.; Starr, A.; Vexler, A.; Karaush, V.; Loew, V.; Greif, J.; Fenig, E.; Aderka, D.; Ben-Yosef, R. Inhibition of pancreatic and lung adenocarcinoma cell survival by curcumin is associated with increased apoptosis, down-regulation of COX-2 and EGFR and inhibition of Erk1/2 activity. Anticancer Res. 2006, 26, 4423–4430.
- Nautiyal, J.; Banerjee, S.; Kanwar, S.S.; Yu, Y.; Patel, B.B.; Sarkar, F.H.; Majumdar, A.P.N. Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. Int. J. Cancer 2011, 128, 951–961.
- Chen, A.; Xu, J.; Johnson, A.C. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr1. Oncogene 2006, 25, 278–287.
- Chen, A.; Xu, J. Activation of PPAR by curcumin inhibits Moser cell growth and mediates suppression of gene expression of cyclin D1 and EGFR. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G447–G456.
- Kane, A.M.; Liu, C.; Akhter, D.T.; McKeone, D.M.; Bell, C.A.; Thurecht, K.J.; Leggett, B.A.; Whitehall, V.L.J. Curcumin chemoprevention reduces the incidence of Braf mutant colorectal cancer in a preclinical study. Dig. Dis. Sci. 2021, 66, 4326–4332.
- Luo, H.; Umebayashi, M.; Doi, K.; Morisaki, T.; Shirasawa, S.; Tsunoda, T. Resveratrol overcomes cellular resistance to vemurafenib through dephosphorylation of AKT in BRAF-mutant melanoma cells. Anticancer Res. 2016, 36, 3585–3590.
- Lu, M.-D.; Li, H.; Nie, J.-H.; Li, S.; Ye, H.-S.; Li, T.-T.; Wu, M.-L.; Liu, J. Dual inhibition of BRAF-MAPK and STAT3 signaling pathways in resveratrol-suppressed anaplastic thyroid cancer cells with BRAF mutations. Int. J. Mol. Sci. 2022, 23, 14385.
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987, 262, 5592–5595.
- Chae, H.-S.; Xu, R.; Won, J.-Y.; Chin, Y.-W.; Yim, H. Molecular targets of genistein and its related flavonoids to exert anticancer effects. Int. J. Mol. Sci. 2019, 20, 2420.
- Lazarevic, B.; Boezelijn, G.; Diep, L.M.; Kvernrod, K.; Ogren, O.; Ramberg, H.; Moen, A.; Wessel, N.; Berg, R.E.; Egge-Jacobsen, W.; et al. Efficacy and safety of short-term genistein intervention in patients with localized prostate cancer prior to radical prostatectomy: A randomized, placebo-controlled, double-blind Phase 2 clinical trial. Nutr. Cancer 2011, 63, 889–898.
- Yoon, H.-S.; Ramachandiran, S.; Chacko, M.A.S.; Monks, T.J.; Lau, S.S. Tuberous sclerosis-2 tumor suppressor modulates ERK and B-Raf activity in transformed renal epithelial cells. Am. J. Physiol. Renal Physiol. 2004, 286, F417–F424.
- Li, Y.; Yang, G.; Zhang, J.; Tang, P.; Yang, C.; Wang, G.; Chen, J.; Liu, J.; Zhang, L.; Ouyang, L. Discovery, synthesis, and evaluation of highly selective vascular endothelial growth factor receptor 3 (VEGFR3) inhibitor for the potential treatment of metastatic triple-negative breast cancer. J. Med. Chem. 2021, 64, 12022–12048.
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978.
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020, 8, 599281.
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 89.
- Zhang, Y.; Zou, J.-Y.; Wang, Z.; Wang, Y. Fruquintinib: A novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manag. Res. 2019, 11, 7787–7803.
- Wang-Gillam, A.; Schelman, W.; Ukrainskyj, S.; Chien, C.; Gonzalez, M.; Yang, Z.; Kania, M.; Yeckes-Rodin, H. Phase 1/1b open-label, dose-escalation study of fruquintinib in patients with advanced solid tumors in the United States. Investig. New Drugs 2023, 41, 851–860.
- Mahipal, A.; Grothey, A. Role of biologics in first-line treatment of colorectal cancer. J. Oncol. Pract. 2016, 12, 1219–1228.
- Jung, H.; Bae, K.; Lee, J.Y.; Kim, J.-H.; Han, H.-J.; Yoon, H.-Y.; Yoon, K.-A. Establishment of canine transitional cell carcinoma cell lines harboring BRAF V595E mutation as a therapeutic target. Int. J. Mol. Sci. 2021, 22, 9151.
- Broecker-Preuss, M.; Müller, S.; Britten, M.; Worm, K.; Schmid, K.W.; Mann, K.; Fuhrer, D. Sorafenib inhibits intracellular signaling pathways and induces cell cycle arrest and cell death in thyroid carcinoma cells irrespective of histological origin or BRAF mutation status. BMC Cancer 2015, 15, 184.
- Wang, H.; Quan, H.; Lou, L. AKT is critically involved in the antagonism of BRAF inhibitor sorafenib against dabrafenib in colorectal cancer cells harboring both wild-type and mutant (V600E) BRAF genes. Biochem. Biophys. Res. Commun. 2017, 489, 14–20.
- Kim, J.E.; Kim, K.K.; Kim, S.Y.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, S.T. MAP2K1 mutation in colorectal cancer patients: Therapeutic challenge using patient-derived tumor cell lines. J. Cancer 2017, 8, 2263–2268.
- Iglesias-Martinez, L.F.; Rauch, N.; Wynne, K.; McCann, B.; Kolch, W.; Rauch, J. Interactome dynamics of RAF1-BRAF kinase monomers and dimers. Sci. Data 2023, 10, 203.
- Mooz, J.; Oberoi-Khanuja, T.K.; Harms, G.S.; Wang, W.; Jaiswal, B.S.; Seshagiri, S.; Tikkanen, R.; Rajalingam, K. Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci. Signal. 2014, 7, ra73.
- Imielinski, M.; Greulich, H.; Kaplan, B.; Araujo, L.; Amann, J.; Horn, L.; Schiller, J.; Villalona-Calero, M.A.; Meyerson, M.; Carbone, D.P. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J. Clin. Investig. 2014, 124, 1582–1586.
- Botton, T.; Yeh, I.; Nelson, T.; Vemula, S.S.; Sparatta, A.; Garrido, M.C.; Allegra, M.; Rocchi, S.; Bahadoran, P.; McCalmont, T.H.; et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res. 2013, 26, 845–851.
- Mologni, L.; Costanza, M.; Sharma, G.G.; Viltadi, M.; Massimino, L.; Citterio, S.; Purgante, S.; Raman, H.; Pirola, A.; Zucchetti, M.; et al. Concomitant BCORL1 and BRAF mutations in vemurafenib-resistant melanoma cells. Neoplasia 2018, 20, 467–477.
- Mullaguri, S.C.; Akula, S.; Ashireddygari, V.R.; Sahoo, P.S.; Burra, V.L.S.P.; Silveri, R.; Mupparapu, V.; Korikani, M.; Amanchi, N.R.; Subramaniam, J.; et al. Estimated sensitivity profiles of lung cancer specific uncommon BRAF mutants towards experimental and clinically approved kinase inhibitors. Toxicol. Appl. Pharmacol. 2022, 453, 116213.
- Molnár, E.; Rittler, D.; Baranyi, M.; Grusch, M.; Berger, W.; Döme, B.; Tóvári, J.; Aigner, C.; Timár, J.; Garay, T.; et al. Pan-RAF and MEK vertical inhibition enhances therapeutic response in non-V600 BRAF mutant cells. BMC Cancer 2018, 18, 542.
- Nagaria, T.S.; Williams, J.L.; Leduc, C.; Squire, J.A.; Greer, P.A.; Sangrar, W. Flavopiridol synergizes with sorafenib to induce cytotoxicity and potentiate antitumorigenic activity in EGFR/HER-2 and mutant RAS/RAF breast cancer model systems. Neoplasia 2013, 15, 939–951.
- Hilhorst, R.; van den Berg, A.; Boender, P.; van Wezel, T.; Kievits, T.; de Wijn, R.; Ruijtenbeek, R.; Corver, W.E.; Morreau, H. Differentiating benign from malignant thyroid tumors by kinase activity profiling and dabrafenib BRAF V600E targeting. Cancers 2023, 15, 4477.
- Ricci, M.S.; Kim, S.-H.; Ogi, K.; Plastaras, J.P.; Ling, J.; Wang, W.; Jin, Z.; Liu, Y.Y.; Dicker, D.T.; Chiao, P.J.; et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007, 12, 66–80.
- Oikonomuu, E.; Koc, M.; Sourkova, V.; Andera, L.; Pintzas, A. Selective BRAFV600E inhibitor PLX4720, requires TRAIL assistance to overcome oncogenic PIK3CA resistance. PLoS ONE 2011, 6, e21632.
- Martinelli, E.; Troiani, T.; Morgillo, F.; Rodolico, G.; Vitagliano, D.; Morelli, M.P.; Tuccillo, C.; Vecchione, L.; Capasso, A.; Orditura, M.; et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin. Cancer Res. 2010, 16, 4990–5001.
- Piscazzi, A.; Costantino, E.; Maddalena, F.; Natalicchio, M.I.; Gerardi, A.M.T.; Antonetti, R.; Cignarelli, M.; Landriscina, M. Activation of the RAS/RAF/ERK signaling pathway contributes to resistance to sunitinib in thyroid carcinoma cell lines. J. Clin. Endocrinol. Metab. 2012, 97, E898–E906.
- Gril, B.; Palmieri, D.; Qian, Y.; Smart, D.D.; Ileva, L.; Liewehr, D.J.; Steinberg, S.M.; Steeg, P.S. Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2+ breast cancer metastasis. Clin. Cancer Res. 2011, 17, 142–153.
- Gunda, V.; Ghosh, C.; Hu, J.; Zhang, L.; Zhang, Y.-Q.; Shen, M.; Kebebew, E. Combination BRAFV600E inhibition with the multitargeting tyrosine kinase inhibitor axitinib shows additive anticancer activity in BRAFV600E-mutant anaplastic thyroid cancer. Thyroid 2023, 33, 1201–1214.
- Tran, K.T.; Kolekar, S.; Wang, Q.; Shih, J.-H.; Buchanan, C.M.; Deva, S.; Shepherd, P.R. Response to BRAF-targeted therapy is enhanced by cotargeting VEGFRs or WNT/β-catenin signaling in BRAF-mutant colorectal cancer models. Mol. Cancer Ther. 2022, 21, 1777–1787.
- Ghosh, C.; Kumar, S.; Kushchayeva, Y.; Gaskins, K.; Boufraqech, M.; Wei, D.; Gara, S.K.; Zhang, L.; Zhang, Y.; Shen, M.; et al. A combinatorial strategy for targeting BRAFV600E-mmtant cancers with BRAFV600E inhibitor (PLX4720) and tyrosine kinase inhibitor (ponatinib). Clin. Cancer Res. 2020, 26, 2022–2036.
- Mordant, P.; Loriot, Y.; Leteur, C.; Calderaro, J.; Bourhis, J.; Wislez, M.; Soria, J.-C.; Deutsch, E. Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive the activity of RAF265, a novel RAFA/VEGFR2 inhibitor, and RAD001 (everolimus) in combination. Mol. Cancer Ther. 2010, 9, 358–368.
- Chow, A.K.M.; Cheng, N.S.M.; Lam, C.S.C.; Ng, L.; Wong, S.K.M.; Wan, T.M.H.; Man, J.H.W.; Cheung, A.H.K.; Yau, T.C.C.; Poon, J.T.C.; et al. Preclinical analysis of the anti-tumor and anti-metastatic effects of Raf265 on colon cancer cells and CD26+ cancer stem cells in colorectal carcinoma. Mol. Cancer 2015, 14, 80.
- Su, Y.; Vilgelm, A.E.; Kelley, M.C.; Hawkins, O.E.; Liu, Y.; Boyd, K.L.; Kantrow, S.; Splittgerber, R.C.; Short, S.P.; Sobolik, T.; et al. RAF265 inhibits the growth of advanced human melanoma tumors. Clin. Cancer Res. 2012, 18, 2184–2198.
- Barollo, S.; Bertazza, L.; Baldini, E.; Ulisse, S.; Cavedon, E.; Boscaro, M.; Pezzani, R.; Mian, C. The combination of RAF265, SB590885, ZSTK474 on thyroid cancer cell lines deeply impact on proliferation and MAPK and PI3K/Akt signaling pathways. Investig. New Drugs 2014, 32, 626–635.
- Saleh, K.; Al Sakhen, M.; Kanaan, S.; Yasin, S.; Höpfner, M.; Tahtamouni, L.; Biersack, B. Antitumor activity of the new tyrphostin briva against BRAFV600E-mutant colorectal carcinoma cells. Investig. New Drugs 2023, 41, 791–801.
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Cappellini, G.C.A.; D’Atri, S. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafinib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174.
- Caporali, S.; Amaro, A.; Levati, L.; Alvino, E.; Lacal, P.M.; Mastroeni, S.; Ruffini, F.; Bonmassar, L.; Cappellini, G.C.A.; Felli, N.; et al. miR-126-3p down-regulation contributes to dabrafenib acquired resistance in melanoma by up-regulating ADAM9 and VEGF-A. J. Exp. Clin. Cancer Res. 2019, 38, 272.
- Martin, M.J.; Hayward, R.; Viros, A.; Marais, R. Metformin accelerates the growth of BRAFV600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012, 2, 344–355.
- Kurenova, E.; Ucar, D.; Liao, J.; Yemma, M.; Gogate, P.; Bshara, W.; Sunar, U.; Seshadri, M.; Hochwald, S.N.; Cance, W.G. A FAK scaffold inhibitor disrupts FAK and VEGFR-3 signaling and blocks melanoma growth by targeting both tumor and endothelial cells. Cell Cycle 2014, 13, 2542–2553.
- Coupe, N.; Guo, L.; Bridges, E.; Campo, L.; Espinosa, O.; Colling, R.; Marshall, A.; Nandakumar, A.; van Stiphout, R.; Buffa, F.M.; et al. WNT5A-ROR2 axis mediates VEGF dependence of BRAF mutant melanoma. Cell. Oncol. 2023, 46, 391–407.
- Ott, P.A.; Hamilton, A.; Min, C.; Safarzadeh-Amiri, S.; Goldberg, L.; Yoon, J.; Yee, H.; Buckley, M.; Christos, P.J.; Wright, J.J.; et al. A phase II trial of sorafenib in metastatic melanoma with tissue correlates. PLoS ONE 2010, 5, e15588.
- Wilson, M.A.; Zhao, F.; Letrero, R.; D’Andrea, K.; Rimm, D.L.; Kirkwood, J.M.; Kluger, H.M.; Lee, S.J.; Schuchter, L.M.; Flaherty, K.T.; et al. Correlation of somatic mutations and clinical outcome in melanoma patients treated with carboplatin, paclitaxel, and sorafenib. Clin. Cancer Res. 2014, 20, 3328–3337.
- Al-Marrawi, M.Y.; Saroya, B.S.; Brennan, M.C.; Yang, Z.; Dykes, T.M.; El-Deiry, W.S. Off-label use of cetuximab plus sorafenib and panitumumab plus regorafenib to personalize therapy for a patient with V600E BRAF-mutant metastatic colon cancer. Cancer Biol. Ther. 2013, 15, 703–710.
- Janku, F.; Sakamuri, D.; Kato, S.; Huang, H.J.; Call, G.; Naing, A.; Holley, V.R.; Patel, S.P.; Amaria, R.N.; Falchook, G.S.; et al. Dose-escalation study of vemurafenib with sorafenib or critozinib in patients with BRAF-mutated advanced cancers. Cancer 2021, 127, 391–402.
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312.
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629.
- Matsumoto, T.; Ikoma, T.; Yamamura, S.; Miura, K.; Tsukidi, T.; Watanabe, T.; Nagai, H.; Takatani, M.; Yasui, H. Regorafenib is suitable for advanced colorectal cancer patients who have previously received trifluridine/tipiracil plus bevacizumab. Sci. Rep. 2023, 13, 2433.
- Izar, B.; Sharfman, W.; Hodi, F.S.; Lawrence, D.; Flaherty, K.T.; Amaravadi, R.; Kim, K.B.; Puzanov, I.; Sosman, J.; Dummer, R.; et al. A first-in-human phase I, multicenter, open-label, dose-escalation study of the oral RAF/VEGFR-2 inhibitor (RAF265) in locally advanced or metastatic melanoma independent from BRAF mutation status. Cancer Med. 2017, 6, 1904–1914.
- Xu, T.; Li, J.; Wang, Z.; Zhang, X.; Zhou, J.; Lu, Z.; Shen, L.; Wang, X. Real-world treatment and outcomes of patients with metastatic BRAF mutant colorectal cancer. Cancer Med. 2023, 12, 10473–10484.
- Stintzing, S.; Heinrich, K.; Tougeron, D.; Modest, D.P.; Schwaner, I.; Eucker, J.; Pihusch, R.; Stauch, M.; Kaiser, F.; Kahl, C.; et al. FOLFOXIRI plus cetuximab or bevacizumab as first-line treatment of BRAFV600E-mutant metastatic colorectal cancer: The randomized phase II FIRE-4.5 (AIO KRK0116) study. J. Clin. Oncol. 2023, 41, 4143–4153.
- Gelsomino, F.; Casadei-Gardini, A.; Rossini, D.; Boccaccino, A.; Masi, G.; Cremolini, C.; Spallanzani, A.; Viola, M.G.; Garajovà, I.; Salati, M.; et al. The role of anti-angiogenics in pre-treated metastatic BRAF-mutant colorectal cancer: A pooled analysis. Cancers 2020, 12, 1022.
- Kang, S.; Lee, M.-W.; Song, I.-C.; Lee, H.-J.; Yun, H.-J.; Jo, D.-Y.; Kim, J.S.; Kwon, J.H.; Kim, J.-Y.; Lee, K.-H.; et al. Maintenance therapy with fluoropyrimidine and cetuximab or bevacizumab after first line FOLFOX-chemotherapy in metastatic colorectal cancer according to RAS or BRAFV600E mutation status. J. Cancer Res. Clin. Oncol. 2023, 149, 7819–7829.
- Yoshino, T.; Portnoy, D.C.; Obermannová, R.; Bodoky, G.; Prausová, J.; Garcia-Carbonero, R.; Ciuleanu, T.; García-Alfonso, P.; Cohn, A.L.; Van Cutsem, E.; et al. Biomarker analysis beyond angiogenesis: RAS/RAF mutation status, tumour sidedness, and second-line ramucirumab efficacy in patients with metastatic colorectal carcinoma from RAISE-a global phase III study. Ann. Oncol. 2019, 30, 124–131.
- Astorga, B.G.; Ballabrera, F.S.; Aguilar, E.A.; Fernández, E.É.; García-Alfonso, P.; Flores, E.G.; García, R.V.; Montes, A.E.; Munoz, A.M.L.; Salvia, A.S. Patient profiles as an aim to optimize selection in the second line setting: The role of aflibercept. Clin. Transl. Oncol. 2021, 23, 1520–1528.
- El-Deiry, W.S.; Winer, A.; Slifker, M.; Taylor, S.; Adamson, B.J.S.; Meropol, N.J.; Ross, E.A. Disease control with FOLFIRI plus ziv-aflibercept (zFOLFIRI) beyond FOLFIRI plus bevacizumab: Case series in metastatic colorectal cancer (mCRC). Front. Oncol. 2019, 9, 142.
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor. Rev. 2004, 15, 197–204.
- Valius, M.; Kazlauskas, A. Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell 1993, 73, 321–334.
- Lassus, H.; Sihto, H.; Leminen, A.; Nordling, S.; Joensuu, H.; Nupponen, N.N.; Butzow, R. Genetic alterations and protein expression of KIT and PDGFRA in serous ovarian carcinoma. Br. J. Cancer 2004, 91, 2048–2055.
- Blom, T.; Roselli, A.; Häyry, V.; Tynninen, O.; Wartiovaara, K.; Korja, M.; Nordfors, K.; Haapasalo, H.; Nupponen, N.N. Amplification and overexpression of KIT, PDGFRA, and VEGFR2 in medulloblastomas and primitive neuroectodermal tumors. J. Neurooncol. 2010, 97, 217–224.
- Tsao, A.S.; Wei, W.; Kuhn, E.; Spencer, L.; Solis, L.M.; Suraokar, M.; Lee, J.J.; Hong, W.K.; Wistuba, I.I. Immunohistochemical overexpression of platelet-derived growth factor receptor-beta (PDGFR-β) is associated with PDGFRB gene copy number gain in sarcomatoid non-small-cell lung cancer. Clin. Lung Cancer 2011, 12, 369–374.
- Ong, H.S.; Gokavarapu, S.; Tian, Z.; Li, J.; Xu, Q.; Cao, W.; Zhang, C.P. PDGFRA mRNA is overexpressed in oral cancer patients as compared to normal subjects with a significant trend of overexpression among tobacco users. J. Oral Pathol. Med. 2017, 46, 591–597.
- Ong, H.S.; Gokavarapu, S.; Tian, Z.; Li, J.; Xu, Q.; Zhang, C.P.; Cao, W. PDGFRA mRNA overexpression is associated with regional metastasis and reduced survival in oral squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 652–659.
- Penzel, R.; Aulmann, S.; Moock, M.; Schwarzbach, M.; Rieker, R.J.; Mechtersheimer, G. The location of KIT and PDGFRA gene mutations in gastrointestinal stromal tumours is site and phenotype associated. J. Clin. Pathol. 2005, 58, 634–639.
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102.
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710.
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol 3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54.
- Joensuu, H.; Rutkowski, P.; Nishida, T.; Steigen, S.E.; Brabec, P.; Plank, L.; Nilsson, B.; Braconi, C.; Bordoni, A.; Magnusson, M.K.; et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J. Clin. Oncol. 2015, 33, 634–642.
- Velghe, A.I.; Van Cauwenberghe, S.; Polyansky, A.A.; Chand, D.; Montano-Almendras, C.P.; Charni, S.; Hallberg, B.; Essaghir, A.; Demoulin, J.-B. PDGFRA alterations in cancer: Characterization of a gain-of-function V536E transmembrane mutant as well as loss-of-function and passenger mutations. Oncogene 2014, 33, 2568–2576.
- Stover, E.H.; Chen, J.; Folens, C.; Lee, B.H.; Mentens, N.; Marynen, P.; Williams, I.R.; Gilliland, D.G.; Cools, J. Activation of FIP1L1-PDGFRalpha requires disruption of the juxtamembrane domain of PDGFRalpha and is FIP1L1-independent. Proc. Natl. Acad. Sci. USA 2006, 103, 8078–8083.
- Campregher, P.V.; Halley, N.D.S.; Vieira, G.A.; Fernandes, J.F.; Velloso, E.D.R.P.; Ali, S.; Mughal, T.; Miller, V.; Mangueira, C.L.P.; Odone, V.; et al. Identification of a novel fusion TBL1XR1-PDGFRB in a patient with acute myeloid leukemia harboring the DEK-Nup214 fusion and clinical response to dasatinib. Leuk. Lymphoma 2017, 58, 2969–2972.
- Sheng, G.; Zeng, Z.; Pan, J.; Kou, L.; Wang, Q.; Yao, H.; Wen, L.; Ma, L.; Wu, D.; Qiu, H.; et al. Multiple MYO18A-PDGFRB fusion transcripts in a myeloproliferative neoplasm patient with T(5;17)(q32;q11). Mol. Cytogenet. 2017, 10, 4.
- Matei, D.; Emerson, R.E.; Lai, Y.C.; Baldridge, L.A.; Rao, J.; Yiannoutsos, C.; Donner, D.D. Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene 2006, 25, 2060–2069.
- Adewuyi, E.E.; Deschenes, J.; Lopez-Campistrous, A.; Kattar, M.M.; Ghosh, S.; McMullen, T.P.W. Autocrine activation of platelet-derived growth factor receptor α in metastatic papillary thyroid cancer. Hum. Pathol. 2018, 75, 146–153.
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735.
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570.
- Oliveira, S.; Lukacs, N. Stem cell factor: A hemopoietic cytokine with important targets in asthma. Curr. Drug Targets Inflamm. Allergy 2003, 2, 313–318.
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564.
- Beghini, A.; Peterlongo, P.; Ripamonti, C.B.; Larizza, L.; Cairoli, R.; Morra, E.; Mecucci, C. c-kit mutations in core binding factor leukemias. Blood 2000, 95, 726–728.
- Louveau, B.; Jouenne, F.; de Moura, C.R.; Sadoux, A.; Baroudjian, B.; Delyon, J.; Herms, F.; De Masson, A.; Da Meda, L.; Battistella, M.; et al. Baseline genomic features in BRAFV600-mutated metastatic melanoma patients treated with BRAF inhibitor + MEK inhibitor in routine care. Cancers 2019, 11, 1203.
- Hongyo, T.; Li, T.; Syaifudin, M.; Baskar, R.; Ikeda, H.; Kanakura, Y.; Aozasa, K.; Nomura, T. Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res. 2000, 60, 2345–2347.
- Büttner, C.; Henz, B.M.; Welker, P.; Sepp, N.T.; Grabbe, J. Identification of activating c-kit mutations in adult-, but not in childhood-onset indolent mastocytosis: A possible explanation for divergent clinical behavior. J. Investig. Dermatol. 1998, 111, 1227–1231.
- Ashman, L.K.; Ferrao, P.; Cole, S.R.; Cambareri, A.C. Effects of mutant c-kit in early myeloid cells. Leuk. Lymphoma 1999, 34, 451–461.
- Babaei, M.A.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459.
- Jonas, O.; Oudin, M.J.; Kosciuk, T.; Whitman, M.; Gertler, F.B.; Cima, M.J.; Flaherty, K.T.; Langer, R. Parallel in-vivo assessment of drug phenotypes at various time points during systemic BRAF inhibition reveals tumor adaption and altered treatment vulnerabilities. Clin. Cancer Res. 2016, 22, 6031–6038.
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to BRAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977.
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011, 71, 5067–5074.
- Rebecca, V.W.; Wood, E.; Fedorenko, I.V.; Paraiso, K.H.T.; Haarberg, H.E.; Chen, Y.; Xiang, Y.; Sarnaik, A.; Gibney, G.T.; Sondak, V.K.; et al. Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Mol. Cell. Proteom. 2014, 13, 1844–1854.
- Vella, L.J.; Behren, A.; Coleman, B.; Greening, D.W.; Hill, A.F.; Cebon, J. Intercellular resistance to BRAF inhibition can be mediated by extracellular vesicle-asssociated PDGFRβ. Neoplasia 2017, 19, 932–940.
- Shi, H.; Hong, A.; Kong, X.; Koya, R.C.; Song, C.; Moriceau, G.; Hugo, W.; Yu, C.C.; Ng, C.; Chodon, T.; et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014, 4, 69–79.
- Adam, C.; Fusi, L.; Weiss, N.; Goller, S.G.; Meder, K.; Frings, V.G.; Kneitz, H.; Goebeler, M.; Houben, R.; Schrama, D.; et al. Efficient suppression of NRAS-driven melanoma by co-inhibition of ERK1/2 and ERK5 MAPK pathways. J. Investig. Dermatol. 2020, 140, 2455–2465.
- Guida, T.; Anaganti, S.; Provitera, L.; Gedrich, R.; Sullivan, E.; Wilhelm, S.M.; Santoro, M.; Carlomagno, F. Sorafenib inhibits imatinib-resistant KIT and platelet-derived growth factor receptor beta gatekeeper mutants. Clin. Cancer Res. 2007, 13, 3363–3369.
- Tiago, M.; Capparelli, C.; Erkes, D.A.; Purwin, T.J.; Heilman, S.A.; Berger, A.C.; Davies, M.A.; Aplin, A.E. Targeting BRD/BET proteins inhibits adaptive kinome upregulation and enhances the effects of BRAF/MEK inhibitors in melanoma. Br. J. Cancer 2020, 122, 789–800.
- Sabbatino, F.; Wang, Y.; Wang, X.; Flaherty, K.T.; Yu, L.; Pepin, D.; Scognamiglio, G.; Pepe, S.; Kirkwood, J.M.; Cooper, Z.A.; et al. PDGFRa up-regulation mediated by Sonic Hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget 2014, 5, 1926–1941.
- Li, Y.; Li, Y.; Liu, Q.; Wang, A. Typrhostin AG1296, a platelet-derived growth factor receptor inhibitor, induces apoptosis, and reduces viability and migration of PLX4032-resistant melanoma cells. OncoTargets Ther. 2015, 8, 1043–1051.
- Che, H.; Guo, H.; Si, X.; You, Q.; Lou, W. Additive effect by combination of Akt inhibitor, MK-2206, and PDGFR inhibitor, tyrphostin AG 1296, in suppressing anaplastic thyroid carcinoma cell viability and motility. OncoTargets Ther. 2014, 7, 425–432.
- Friedman, A.A.; Amzallag, A.; Pruteanu-Malinici, I.; Baniya, S.; Cooper, Z.A.; Piris, A.; Hargreaves, L.; Igras, V.; Frederick, D.T.; Lawrence, D.P.; et al. Landscape of targeted anti-cancer drug synergies in melanoma identifies a novel BRAF-VEGFR/PDGFR combination treatment. PLoS ONE 2015, 10, e0140310.
- Recagni, M.; Tassinari, M.; Doria, F.; Cimino-Reale, G.; Zaffaroni, N.; Freccero, M.; Folini, M.; Richter, S.N. The oncogenic signaling pathways in BRAF-mutant melanoma cells are modulated by naphthalene diimide-like G-quadruple ligands. Cells 2019, 8, 1274.
- Singleton, K.R.; Crawford, L.; Tsui, E.; Manchester, H.E.; Maertens, O.; Liu, X.; Liberti, M.V.; Magpusao, A.N.; Stein, E.M.; Tingley, J.P.; et al. Melanoma therapeutic strategies that select against resistance by exploiting MYC-driven evolutionary convergence. Cell Rep. 2017, 21, 2796–2812.
- Ablain, J.; Liu, S.; Moriceau, G.; Lo, R.S.; Zon, L.I. SPRED1 deletion confers resistance to MAPK inhibition in melanoma. J. Exp. Med. 2020, 218, e20201097.
- Guo, T.; Agaram, N.P.; Wong, G.C.; Hom, G.; D’Alamo, D.; Maki, R.G.; Schwartz, G.K.; Veach, D.; Clarkson, B.D.; Singer, S.; et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin. Cancer Res. 2007, 13, 4874–4881.
- Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 1769–1776.
- Menzer, C.; Hassel, J.C. Targeted therapy for melanomas without BRAF V600 mutations. Curr. Treat. Options Oncol. 2022, 23, 831–842.
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell 2019, 35, 738–751.
- Golcic, M.; Jones, R.L.; Huang, P.; Napolitano, A. Evaluation of systemic treatment options for gastrointestinal stromal tumours. Cancers 2023, 15, 4081.
- Franck, C.; Rosania, R.; Franke, S.; Haybaeck, J.; Canbay, A.; Venerito, M. The BRAF status may predict response to sorafenib in gastrointestinal stromal tumors resistant to imatinib, sunitinib, and regorafenib: Case series and review of the literature. Digestion 2019, 99, 179–184.
- Gelderblom, H.; Jones, R.L.; Blay, J.-Y.; George, S.; von Mehren, M.; Zalcberg, J.R.; Kng, Y.-K.; Razak, A.A.; Trent, J.; Attia, S.; et al. Patient-reported outcomes and tolerability in patients receiving ripretinib versus sunitinib after treatment with imatinib in INTRIGUE, a phase 3, open-label study. Eur. J. Cancer 2023, 192, 113245.
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF mutant gastrointestinal stromal tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315.
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib: An international, multicentre, prospective, randomised, placebocontrolled phase 3 trial (GRID). Lancet 2013, 381, 295–302.
- Nannini, M.; Valerio, D.S.; Gruppioni, E.; Altimari, A.; Chiusole, B.; Saponara, M.; Pantaleo, M.A.; Brunello, A. Complete radiological response to first-line regorafenib in a patient with abdominal relapse of BRAF V600E mutated GIST. Ther. Adv. Gastroenterol. 2020, 13, 1–4.
- Martin-Broto, J.; Valverde, C.; Hindi, N.; Vincenzi, B.; Martinez-Trufero, J.; Grignani, G.; Italiano, A.; Lavernia, J.; Vallejo, A.; Dei Tos, P.; et al. REGISTRI: Regorafenib in first-line of KIT/PDGFRA wild type metastatic GIST: A collaborative Spanish (GEIS), Italian (ISG) and French Sarcoma Group (FSG) phase II trial. Mol. Cancer 2023, 22, 127.
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892.
- Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-dependent molecular mechanisms underlying anti-cancer drug resistance. Cancers 2021, 13, 5796.
- Metzner, T.; Bedeir, A.; Held, G.; Peter-Vörösmarty, B.; Ghassemi, S.; Heinzle, C.; Spiegl-Kreinecker, S.; Marian, B.; Holzmann, K.; Grasl-Kraupp, B.; et al. Fibroblast growth factor receptors as therapeutic targets in human melanoma: Synergism with BRAF inhibition. J. Investig. Dermatol. 2011, 131, 2087–2095.
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF inhibition causes resilience of melanoma cell lines by inducing the secretion of FGF1. Oncogenesis 2018, 7, 71.
- Ghassemi, S.; Vejdovszky, K.; Sahin, E.; Ratzinger, L.; Schelch, K.; Mohr, T.; Peter-Vörösmarty, B.; Brankovic, J.; Lackner, A.; Leopoldi, A.; et al. FGF5 is expressed in melanoma and enhances malignancy in vitro and in vivo. Oncotarget 2017, 8, 87750–87762.
- Tassone, E.; Valacca, C.; Mignatti, P. Membrane-type 1 matrix metalloproteinase downregulates fibroblast growth factor-2 binding to the cell surface and intracellular signaling. J. Cell. Physiol. 2015, 230, 366–377.
- Garay, T.; Molnár, E.; Juhász, É.; László, V.; Barbai, T.; Dobos, J.; Schelch, K.; Pirker, C.; Grusch, M.; Berger, W.; et al. Sensitivity of melanoma cells to EGFR and FGFR activation but not inhibition is influenced by oncogenic BRAF and NRAS mutations. Pathol. Oncol. Res. 2015, 21, 957–968.
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.-Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.-B. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem. 2012, 287, 28087–28098.
- Lee, C.-K.; Lee, M.-E.; Lee, W.S.; Kim, J.M.; Park, K.H.; Kim, T.S.; Lee, K.Y.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Dovitinib (TKI258), a multi-target angiokinase inhibitor, is effective regardless of KRAS and BRAF mutation status in colorectal cancer. Am. J. Cancer Res. 2015, 5, 72–86.
- Langdon, C.G.; Held, M.A.; Platt, J.T.; Meeth, K.; Iyidogan, P.; Mamillapalli, R.; Koo, A.B.; Klein, M.; Liu, Z.; Bosenberg, M.W.; et al. The broad spectrum receptor tyrosine kinase inhibitor dovitinib suppresses growth of BRAF mutant melanoma cells in combination with other signaling pathway inhibitors. Pigment Cell Melanoma Res. 2015, 28, 417–430.
- Kane, A.M.; Liu, C.; Fennell, L.J.; McKeone, D.M.; Bond, C.E.; Pollock, P.M.; Young, G.; Leggett, B.A.; Whitehall, V.L.J. Aspirin reduces the incidence of metastasis in a pre-clinical study of Braf mutant serrated colorectal neoplasia. Br. J. Cancer 2021, 124, 1820–1827.
- Eigner, K.; Filik, Y.; Mark, F.; Schütz, B.; Klambauer, G.; Moriggl, R.; Hengstschläger, M.; Stangl, H.; Mikula, M.; Röhrl, C. The unfolded protein response impacts melanoma progression by enhancing FGF expression and can be antagonized by a chemical chaperone. Sci. Rep. 2017, 7, 17498.
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target Ther. 2020, 5, 181.
- Czyz, M. Fibroblast growth factor receptor signaling in skin cancers. Cells 2019, 8, 540.
- Kumar, R.; Jain, A.G.; Rashid, M.U.; Ali, S.; Khetpal, N.; Hussain, I.; Ahmad, S. HGFR and FGR2: Their roles in progression and metastasis of esophageal cancer. In Role of Tyrosine Kinases in Gastrointestinal Malignancies; Nagaraju, G.P., Ed.; Springer: Singapore, 2018; pp. 1–14.
- Gumustekin, M.; Kargi, A.; Bulut, G.; Gozukizil, A.; Ulukus, C.; Oztop, I.; Atabey, N. HGF/c-Met overexpressions, but not met mutation, correlates with progression of non-small cell lung cancer. Pathol. Oncol. Res. 2012, 18, 209–218.
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J. Clin. Oncol. 2016, 34, 721–730.
- Zhao, X.; Qu, J.; Hui, Y.; Zhang, H.; Sun, Y.; Liu, X.; Zhao, X.; Zhao, Z.; Yang, Q.; Wang, F.; et al. Clinicopathological and prognostic significance of c-Met overexpression in breast cancer. Oncotarget 2017, 8, 56758–56767.
- Blanc-Durand, F.; Alameddine, R.; Iafrate, A.J.; Tran-Thanh, D.; Lo, Y.C.; Blais, N.; Routy, B.; Tehfé, M.; Leduc, C.; Romeo, P.; et al. Tepotinib efficacy in a patient with non-small cell lung cancer with brain metastasis harboring an HLA-DRB1-MET gene fusion. Oncologist 2020, 25, 916–920.
- Davies, K.D.; Ng, T.L.; Estrada-Bernal, A.; Le, A.T.; Ennever, P.R.; Camidge, D.R.; Doebele, R.C.; Aisner, D.L. Dramatic response to crizotinib in a patient with lung cancer positive for an HLA-DRB1-MET gene fusion. JCO Precis. Oncol. 2017, 1, 1–6.
- Zhu, Y.C.; Wang, W.X.; Song, Z.B.; Zhang, Q.X.; Xu, C.W.; Chen, G.; Zhuang, W.; Lv, T.; Song, Y. MET-UBE2H fusion as a novel mechanism of acquired EGFR resistance in lung adenocarcinoma. J. Thorac. Oncol. 2018, 13, e202–e204.
- Karagonlar, Z.F.; Koc, D.; Iscan, E.; Erdal, E.; Atabey, N. Elevated hepatocyte growth factor expression as an autocrine c-Met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci. 2016, 107, 407–416.
- Horiguchi, N.; Takayama, H.; Toyoda, M.; Otsuka, T.; Fukusato, T.; Merlino, G.; Takagi, H.; Mori, M. Hepatocyte growth factor promotes hepatocarcinogenesis through c-Met autocrine activation and enhanced angiogenesis in transgenic mice treated with diethylnitrosamine. Oncogene 2002, 21, 1791–1799.
- Rasola, A.; Fassetta, M.; De Bacco, F.; D’Alessandro, L.; Gramaglia, D.; Di Renzo, M.F.; Comoglio, P.M. A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene 2007, 26, 1078–1087.
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.M.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122.
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell. Biol. 2010, 11, 834–848.
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Kwon, H.J.; Chang, J.W.; Ban, M.J.; Kim, W.S.; Shin, D.Y.; Lee, E.J.; Koh, Y.W.; et al. c-Met-mediated reactivation of PI3K/AKT signaling contributes to insensitive of BRAF(V600E) mutant thyroid cancer to BRAF inhibition. Mol. Carcinogen. 2016, 55, 1678–1687.
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Ko, S.; Yoon, S.O.; Ku, M.; Yang, J.; Kim, J.W.; Ban, M.J.; Kim, J.-H.; et al. Acquired resistance to BRAF inhibition induces epithelial-to-mesenchymal transition in BRAF (V600E) mutant thyroid cancer by c-Met-mediated AKT activation. Oncotarget 2017, 8, 596–609.
- Knauf, J.A.; Luckett, K.A.; Chen, K.-Y.; Voza, F.; Socci, N.D.; Ghossein, R.; Fagin, J.A. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J. Clin. Investig. 2018, 128, 4086–4097.
- Vergani, E.; Vallacchi, V.; Frigerio, S.; Deho, P.; Mondellini, P.; Perego, P.; Cassinelli, G.; Lanzi, C.; Testi, M.A.; Rivoltini, L.; et al. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia 2011, 13, 1132–1142.
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumor microenvironment induces innate RAF-inhibitor resistance through HFG secretion. Nature 2012, 487, 500–504.
- Caenepeel, S.; Cooke, K.; Wadsworth, S.; Huang, G.; Robert, L.; Moreno, B.H.; Parisi, G.; Cajulis, E.; Kendall, R.; Beltran, P.; et al. MAPK pathway inhibition induces MET and GAB1 levels, priming BRAF mutant melanoma for rescue by hepatocyte growth factor. Oncotarget 2017, 8, 17795–17809.
- Das, I.; Wilhelm, M.; Hölom, V.; Marquez, R.F.; Svedman, F.C.; Hansson, J.; Tuominen, R.; Brage, S.E. Combining ERBB family and MET inhibitors is an effective therapeutic strategy in cutaneous malignant melanoma independent of BRAF/NRAS mutation status. Cell Death Dis. 2019, 10, 663.
- Carson, R.; Celtikci, B.; Fenning, C.; Javadi, A.; Crawford, N.; Carbonell, L.P.; Lawler, M.; Longley, D.B.; Johnston, P.G.; Van Schaeybroeck, S. HDAC inhibition overcomes acute resistance to MEK inhibition in BRAF mutant colorectal cancer by down-regulation of c-FLIPL. Clin. Cancer Res. 2015, 21, 3230–3240.
- Xu, T.; Wang, X.; Wang, Z.; Deng, T.; Qi, C.; Liu, D.; Li, Y.; Ji, C.; Shen, L. Molecular mechanisms underlying the resistance of BRAF V600E-mutant metastatic colorectal cancer to EGFR/BRAF inhibitors. Ther. Adv. Med. Oncol. 2022, 14, 1–12.
- Oddo, D.; Siravegna, G.; Gloghini, A.; Vernieri, C.; Mussolin, B.; Morano, F.; Crisafulli, G.; Berenato, R.; Corti, G.; Volpi, C.C.; et al. Emergence of MET hyper-amplification at progression to MET and BRAF inhibition in colorectal cancer. Br. J. Cancer 2017, 117, 347–352.
- Dong, Y.; Xu, J.; Sun, B.; Wang, J.; Wang, Z. MET-targeted therapies and clinical outcomes. A systematic literature review. Mol. Diagn. Ther. 2022, 26, 203–227.
- Li, S.; Li, X.; Sun, S.; Li, S.; Zhou, C. Response to osimertinib plus trametinib in a heavily treated epidermal growth factor receptor (EGFR)-positive NSCLC harboring a rare, acquired rapidly accelerated fibrosarcoma B-type (BRAF) p.D594N mutation: A case report. Anticancer Drugs 2022, 33, 963–965.
- Recondo, G.; Bahcall, M.; Spurr, L.F.; Che, J.; Ricciuti, B.; Leonardi, G.C.; Lo, Y.-C.; Li, Y.Y.; Lamberti, G.; Nguyen, T.; et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14-mutant NSCLC. Clin. Cancer Res. 2020, 26, 2615–2625.
- Chen, H.X.; Sharon, E. IGF-1R as an anticancer target—Trials and tribulations. Chin. J. Cancer 2013, 32, 242–252.
- Hakuno, F.; Takahashi, S.-I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86.
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423.
- Rasmussen, A.A.; Cullen, K.J. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. Breast Cancer Res. Treat. 1998, 47, 219–233.
- Pavelić, J.; Radaković, B.; Pavelić, K. Insulin-like growth factor 2 and its receptors (IGF 1R and IGF 2R/mannose 6-phosphate) in endometrial adenocarcinoma. Gynecol. Oncol. 2007, 105, 727–735.
- Zheng, X.; Lu, G.; Yao, Y.; Gu, W. An autocrine IL-6/IGF-1R loop mediates EMT and promotes tumor growth in non-small cell lung cancer. Int. J. Biol. Sci. 2019, 15, 1882–1891.
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695.
- Gopal, Y.N.V.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010, 70, 8736–8747.
- Benito-Jardón, L.; Díaz-Martínez, M.; Arellano-Sánchez, N.; Vaquero-Morales, P.; Esparís-Ogando, A.; Teixidó, J. Resistance to MAPK inhibitors in melanoma involves activation of the IGF1R-MEK5-Erk5 pathway. Cancer Res. 2019, 79, 2244–2256.
- Flanigan, S.A.; Pitts, T.M.; Newton, T.P.; Kulikowski, G.N.; Tan, A.C.; McManus, M.C.; Spreafico, A.; Kachaeva, M.I.; Selby, H.M.; Tentler, J.J.; et al. Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin. Cancer Res. 2013, 19, 6219–6229.
- Wu, X.; Marmarelis, M.E.; Hodi, F.S. Activity of the heat shock protein 90 inhibitor gantespib in melanoma. PLoS ONE 2013, 8, e56134.
- Olbryt, M.; Rusin, A.; Fokt, I.; Habryka, A.; Tudrej, P.; Student, S.; Sochanik, A.; Zielinski, R.; Priebe, W. Bis-antracycline WP760 abrogates melanoma growth by transcription inhibition, p53 activation and IGF1R downregulation. Investig. New Drugs 2017, 35, 545–555.
- Ramcharan, R.; Aleksic, T.; Kamdoum, W.P.; Gao, S.; Pfister, S.X.; Tanner, J.; Bridges, E.; Asher, R.; Watson, A.J.; Margison, G.P.; et al. IGF-1R inhibition induces schedule-dependent sensitization of human melanoma to temozolomide. Oncotarget 2015, 6, 39877–39890.
- Herkert, B.; Kauffmann, A.; Mollé, S.; Schnell, C.; Ferrat, T.; Voshol, H.; Juengert, J.; Erasimus, H.; Marszalek, G.; Kazic-Legueux, M.; et al. Maximizing the efficacy of MAPK-targeted treatment in PTENLOF/BRAFMUT melanoma through PI3K and IGF1R inhibition. Cancer Res. 2016, 76, 390–402.
- Huether, A.; Höpfner, M.; Baradari, V.; Schuppan, D.; Scherübl, H. Sorafenib alone or as combination therapy for growth control of cholangiocarcinoma. Biochem. Pharmacol. 2007, 73, 1308–1317.
- Bugide, S.; Parajuli, K.R.; Chava, S.; Pattanayak, R.; Della Manna, D.L.; Shrestha, D.; Yang, E.S.; Cai, G.; Johnson, D.B.; Gupta, R. Loss of HAT1 expression confers BRAFV600E inhibitor resistance to melanoma cells by activating MAPK signaling via IGF1R. Oncogenesis 2020, 9, 44.
- Strub, T.; Ghiraldini, F.G.; Carcamo, S.; Li, M.; Wroblewska, A.; Singh, R.; Goldberg, M.S.; Hasson, D.; Wang, Z.; Gallagher, S.J.; et al. SIRT6 haploinsufficiency induces BRAFV600E melanoma cell resistance to MAPK inhibitors via IGF signaling. Nat. Commun. 2018, 9, 3440.
- Werner, H.; Sarfstein, R.; Bruchim, I. Investigational IGF1R inhibitors in early stage clinical trials for cancer therapy. Expert Opin. Invest. Drugs 2019, 28, 1101–1112.
- Wilky, B.A.; Rudek, M.A.; Ahmed, S.; Laheru, D.A.; Cosgrove, D.; Donehower, R.C.; Nelkin, B.; Ball, D.; Doyle, L.A.; Chen, H.; et al. A phase I trial of vertical inhibition of IGF signaling using cixutumumab, and anti-IGF-1R antibody, and selumetinib, an MEK1/2 inhibitor, in advanced solid tumors. Br. J. Cancer 2015, 112, 24–31.
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153.
- Tang, Y.; Zang, H.; Wen, Q.; Fan, S. AXL in cancer: A modulator of drug resistance and therapeutic target. J. Exp. Clin. Cancer Res. 2023, 42, 148.
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289.
- Nyakas, M.; Fleten, K.G.; Haugen, M.H.; Engedal, N.; Sveen, C.; Farstad, I.N.; Florenes, V.A.; Prasmickaite, L.; Maeleandsmo, G.M.; Seip, K. AXL inhibition improves BRAF-targeted treartment in melanoma. Sci. Rep. 2022, 12, 5076.
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827.
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Sharabi, A.; Gresnigt-van den Heuvel, E.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212.
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Pretre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget 2017, 8, 69204–69218.
- Sinik, L.; Minson, K.A.; Tentler, J.J.; Carrico, J.; Bagby, S.M.; Robinson, W.A.; Kami, R.; Burstyn-Cohen, T.; Eckhardt, S.G.; Wang, X.; et al. Inhibition of MERTK promotes suppression of tumor growth in BRAF mutant and BRAF wild-type melanoma. Mol. Cancer Ther. 2019, 18, 278–288.
- Rosen, E.Y.; Won, H.H.; Zheng, Y.; Cocco, E.; Selcuklu, D.; Gong, Y.; Friedman, N.D.; de Bruijn, I.; Sumer, O.; Bielski, C.M.; et al. The evolution of RET inhibitor resistance in RET-driven lung and thyroid cancers. Nat. Commun. 2022, 13, 1450.
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther II, W.; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978.
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariamé, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2) (q25;p23) translocation. Blood 1999, 93, 3088–3095.
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature 2007, 448, 561–566.
- Elliott, J.; Bai, Z.; Hsieh, S.-C.; Kelly, S.E.; Chen, L.; Skidmore, B.; Yousef, S.; Zheng, C.; Stewart, D.J.; Wells, G.A. ALK inhibitors for non-small cell lung cancer: A systematic review and network meta-analysis. PLoS ONE 2020, 15, e0229179.
- Janostiak, R.; Malvi, P.; Wajapeyee, N. Anaplastic lymphoma kinase confers resistance to BRAF kinase inhibitors in melanoma. iScience 2019, 16, 453–467.
- Guo, W.; Liang, J.; Zhang, D.; Huang, X.; Lv, Y. Lung adenocarcinoma harboring complex EML4-ALK fusion and BRAF V600E co-mutation responded to alectenib. Medicine 2022, 101, 40.
- Sui, A.; Song, H.; Li, Y.; Guo, L.; Wang, K.; Yuan, M.; Chen, R. BRAF V600E mutation as a novel mechanisms of acquired resistance to ALK inhibition in ALK-rearranged lung adenocarcinoma—A case report. Medicine 2021, 100, 8.
- Pasau, T.; Wauters, E.; Wauters, I.; Duplaquet, F.; Pirard, L.; Pop-Stanciu, C.; D’Haene, N.; Dupont, M.; Vander Borght, T.; Rondelet, B.; et al. Case report: BRAF A598-T599insV mutation as a potential resistance mechanism to alectinib in ALK-rearranged lung adenocarcinoma. Front. Oncol. 2022, 12, 985446.
- Fu, H.-L.; Valiathan, R.R.; Arkwright, R.; Sohail, A.; Mihai, C.; Kumarasiri, M.; Mahasenan, K.V.; Mobashery, S.; Huang, P.; Agarwal, G.; et al. Discoidin domain receptors: Unique receptor tyrosine kinases in collagen-mediated signaling. J. Biol. Chem. 2013, 288, 7430–7437.
- De Moura, C.R.; Battistella, M.; Sohail, A.; Caudron, A.; Feugeas, J.P.; Podgorniak, M.-P.; Pages, C.; Dorval, S.M.; Marco, O.; Menashi, S.; et al. Discoidin domain receptors: A promising target in melanoma. Pigment Cell Melanoma Res. 2019, 32, 697–707.
- De Moura, C.R.; Prunotto, M.; Sohail, A.; Battistella, M.; Jouenne, F.; Marbach, D.; Lebbé, C.; Fridman, R.; Mourah, S. Discoidin domain receptors in melanoma: Potential therapeutic targets to overcome MAPK inhibitor resistance. Front. Oncol. 2020, 10, 1748.
- Berestjuk, I.; Lecacheur, M.; Carminati, A.; Diazzi, S.; Rovera, C.; Prod’homee, V.; Ohanna, M.; Popovic, A.; Mallavialle, A.; Larbret, F.; et al. Targeting discoidin domain receptors DDR1 and DDR2 overcomes matrix-mediated tumor cell adaption and tolerance to BRAF-targeted therapy in melanoma. EMBO Mol. Med. 2022, 14, e11814.
- Lyon, A.; Tripathi, R.; Meeks, C.; He, D.; Wu, Y.; Liu, J.; Wang, C.; Chen, J.; Zhu, H.; Mukherjee, S.; et al. ABL1/2 and DDR1 drive MEKi resistance in NRAS-mutant melanomas by stabilizing RAF/MYC/ETS1 and promoting RAF homodimerization. Cancers 2023, 15, 954.
- Belli, V.; Napolitano, S.; De Falco, V.; Suarato, G.; Perrone, A.; Guerrera, L.P.; Martini, G.; Della Corte, C.M.; Martinelli, E.; Morgillo, F.; et al. Targeting EphA2 and DDR signaling can overcome BRAF and MEK inhibitors acquired resistance in melanoma cell lines. Transl. Med. Commun. 2023, 8, 3.
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114.
- Peng, Q.; Chen, L.; Wu, W.; Wang, J.; Zheng, X.; Chen, Z.; Jiang, Q.; Han, J.; Wei, L.; Wang, L.; et al. EPH receptor A2 governs a feedback loop that activates Wnt/β-catenin signaling in gastric cancer. Cell Death Dis. 2018, 9, 1146.
- Hamaoka, Y.; Negishi, M.; Katoh, H. EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell Signal. 2016, 28, 937–945.
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495.
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. EphA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015, 5, 274–287.
- Paraiso, K.H.T.; Thakur, M.D.; Fang, B.; Koomen, J.M.; Fedorenko, I.V.; John, J.K.; Tsao, H.; Flaherty, K.T.; Sondak, V.K.; Messina, J.L.; et al. Ligand independent EphA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015, 5, 264–273.
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.-M.; Journe, F. Metabolic reprogramming in metastatic melanoma with acquired resistance to targeted therapies: Integrative metabolomic and proteomic analysis. Cancers 2020, 12, 1323.
- Zhang, C.; Smalley, I.; Emmons, M.F.; Sharma, R.; Izumi, V.; Messina, J.; Koomen, J.M.; Pasquale, E.B.; Forsyth, P.A.; Smalley, K.S.M. Noncanonical EphA2 signaling is a driver of tumor-endothelial cell interactions and metastatic dissemination in BRAF inhibitor-resistant melanoma. J. Investig. Dermatol. 2021, 141, 840–851.
- Azimi, A.; Tuominen, R.; Svedman, F.C.; Caramuta, S.; Pernemalm, M.; Stolt, M.F.; Kanter, L.; Kharaziha, P.; Lehtiö, J.; Johansson, C.H.; et al. Silencing FLI or targeting CD13/ANPEP lead to dephosphorylation of EPHA2, a mediator of BRAF inhibitor resistance, and induce growth arrest or apoptosis in melanoma cells. Cell Death Dis. 2017, 8, e3029.
- Wu, Y.; Huang, J.; Ivan, C.; Sun, Y.; Ma, S.; Mangala, L.S.; Fellman, B.M.; Urbauer, D.L.; Jennings, N.B.; Ram, P.; et al. MEK inhibition overcomes resistance to EphA2-targeted therapy in uterine cancer. Gynecol. Oncol. 2021, 163, 181–190.
- Huang, J.; Hu, W.; Bottsford-Miller, J.; Liu, T.; Han, H.D.; Zand, B.; Pradeep, S.; Roh, J.-W.; Thanapprapasr, D.; Dalton, H.J.; et al. Crosstalk between EphA2 and BRaf/CRaf is a key determinant of response to dasatinib. Clin. Cancer Res. 2014, 20, 1846–1855.
- Annunziata, C.M.; Kohn, E.C.; LoRusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Investig. New Drugs 2013, 31, 77–84.
- Gan, H.K.; Parakh, S.; Lee, F.T.; Tebbutt, N.C.; Ameratunga, M.; Lee, S.T.; O’Keefe, G.J.; Gong, S.J.; Vanrenen, C.; Caine, J.; et al. A phase 1 safety and bioimaging trial of antibody DS-8895a against EphA2 in patients with advanced or metastatic EphA2 positive cancers. Investig. New Drugs 2022, 40, 747–755.
- Sen, B.; Peng, S.; Tang, X.; Erickson, H.S.; Galindo, H.; Mazumdar, T.; Stewart, D.J.; Wistuba, I.; Johnson, F.M. Kinase impaired BRAF mutations confer lung cancer sensitivity to dasatinib. Sci. Transl. Med. 2012, 4, 136ra70.
- Storkus, W.J.; Maurer, D.; Lin, Y.; Ding, F.; Bose, A.; Lowe, D.; Rose, A.; DeMark, M.; Karapetyan, L.; Taylor, J.L.; et al. Dendritic cell vaccines targeting tumor blood vessel antigens in combination with dasatinib induce therapeutic immune responses in patients with checkpoint-refractory advanced melanoma. J. Immunother. Cancer 2021, 9, e003675.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
807
Revisions:
2 times
(View History)
Update Date:
14 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No