Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yeong Ho Kim | -- | 2083 | 2024-03-13 12:14:18 | | | |
| 2 | Wendy Huang | Meta information modification | 2083 | 2024-03-14 05:34:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kim, H.J.; Kim, Y.H. Molecular Pathology of Melanoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/56205 (accessed on 24 June 2026).
Kim HJ, Kim YH. Molecular Pathology of Melanoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/56205. Accessed June 24, 2026.
Kim, Hyun Jee, Yeong Ho Kim. "Molecular Pathology of Melanoma" Encyclopedia, https://encyclopedia.pub/entry/56205 (accessed June 24, 2026).
Kim, H.J., & Kim, Y.H. (2024, March 13). Molecular Pathology of Melanoma. In Encyclopedia. https://encyclopedia.pub/entry/56205
Kim, Hyun Jee and Yeong Ho Kim. "Molecular Pathology of Melanoma." Encyclopedia. Web. 13 March, 2024.
Copy Citation
Melanoma, the deadliest form of skin cancer, presents a significant clinical challenge due to its high metastatic potential and resistance to conventional therapies. It originates from melanocytes and is increasingly prevalent. Melanoma, a highly aggressive skin cancer, is characterized by rapid progression and high mortality. Recent advances in molecular pathogenesis have shed light on genetic and epigenetic changes that drive melanoma development.
melanoma
molecular pathology
genetic mutations
BRAF
NRAS
c-KIT
GNAQ/GNA11
MAPK/ERK pathway
PI3K/AKT/mTOR pathway
melanogenesis
1. Introduction
Melanoma’s incidence surged by 320% from 1975 to 2018, influenced by risk factors such as sun exposure, indoor tanning, family history, and the number of nevi [1]. This type of skin cancer is responsible for nearly 90% of skin cancer deaths, despite constituting a small fraction of skin cancer cases [2]. The disease impacts both older and younger populations, though the increase in incidence is particularly notable among older individuals [2]. The primary cause of death from melanoma is metastatic spread, first to the lymph nodes and, most commonly, to the lungs [2]. Early-stage melanoma (I–II) can be effectively treated with complete surgical excision, boasting an excellent 5-year survival rate of 99.4% [1]. However, the prognosis worsens significantly in advanced stages, with 5-year survival rates dropping to 68% for stage III and 29.8% for stage IV melanoma [1].
The clinical burden of melanoma is growing alongside its rising global incidence, which has been increasing by approximately 3% annually in certain regions [3]. Projections for 2023 estimate 97,620 new cases and 7990 deaths in the United States alone [3]. Meanwhile, the International Agency for Research on Cancer reported an estimated 324,635 new diagnoses and 57,043 deaths from melanoma globally in 2020 [3]. These statistics highlight the critical need for the rational and evidence-based selection of therapies in the treatment of melanoma.
The progression from benign melanocytic nevi to malignant melanoma and metastasis involves an interplay of genetic factors and UV-induced damage. Melanocytic nevi, typically benign, can evolve into melanoma through mutations, primarily BRAFV600E in common nevi and various mutations in MAPK signaling, the TERT promoter, and CDKN2A in dysplastic nevi [4]. The progression is marked by additional mutations, such as in NRAS, and is influenced by the activation of the WNT signaling pathway, which is important for metastasis. Genes such as ARID2 and ARID1A are also implicated in melanoma’s progression [4].
2. WHO Classification and Molecular Diversity of Melanoma
The 2018 WHO classification of melanocytic lesions provides a refined understanding of melanoma’s molecular diversity, categorizing melanomas based on CSD. This system divides melanomas into:
1. Low-CSD melanomas: these include superficial spreading melanomas, typically associated with less sun damage. They often arise on the trunk and proximal areas of the extremities and primarily feature BRAFV600E mutations. Other mutations in these melanomas include the TERT promoter and CDKN2A, with PTEN and TP53 mutations observed in more advanced stages [5].
2. High-CSD melanomas: comprising lentigo maligna and desmoplastic melanomas, these usually develop on heavily sun-damaged skin, particularly in older individuals. They can have a high mutation load, including NRAS, BRAF non-V600E, or NF1 mutations, and they frequently present TERT promoter mutations, CDKN2A, and occasionally KIT mutations. The mutation count in these melanomas increases with the degree of CSD, with desmoplastic melanomas showing the highest tumor mutation burden [5].
3. Non-CSD-associated melanomas: this category contains Spitz melanomas, acral melanomas, mucosal melanomas, melanomas arising from congenital or blue nevi, and uveal melanomas. These subtypes are typically devoid of BRAF, NRAS, and NF1 mutations (triple wild-type), but can feature KIT mutations, gene amplifications, and structural rearrangements, especially of the CCND1 gene and SF3B1. Acral and mucosal melanomas are biologically distinct from their cutaneous counterparts in sun-exposed areas. Spitz melanomas show tyrosine kinase or serine-threonine kinase fusions, and melanomas in blue nevi and uveal melanomas often contain GNA11 or GNAQ mutations [5].
This classification not only segregates melanomas based on CSD, but also correlates the types with specific molecular alterations, providing crucial insights into the varied molecular pathways and risk factors associated with different subtypes of melanoma.
In addition to these classifications, WHO has introduced the concept of „intermediate” lesions in its latest melanocytic tumor classification, acknowledging the diagnostic challenges of melanocytic tumors. This approach shifts away from viewing melanocytic tumors as simply benign or malignant, suggesting a more nuanced understanding by providing nine categories/pathways, each marked by specific genetic drivers [5].
3. Key Genetic Mutations in Melanoma
3.1. BRAF Mutations
BRAF mutations are found in approximately 50% of melanomas and play a crucial role in the disease’s pathogenesis (Figure 1). These mutations are predominantly observed in cutaneous melanomas and are frequently associated with UV radiation exposure. This link is evidenced by the high levels of UV radiation signatures, especially C > T substitutions, found in these tumors [6]. Notably, patients with BRAF mutations tend to be younger than those with other melanoma subtypes [6]. The mutation patterns related to UV exposure highlight the interplay between environmental factors and the genetic landscape of melanoma, emphasizing the importance of considering both of them in understanding melanoma development.
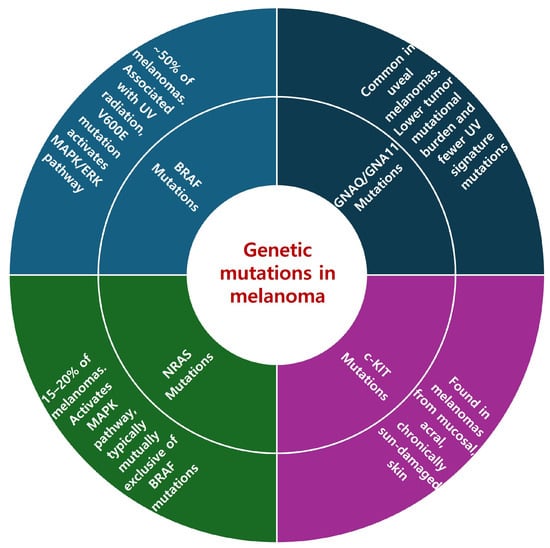
Figure 1. Genetic mutations in melanoma.
The presence of BRAF mutations, particularly V600E, leads to the activation of the MAPK/ERK signaling pathway, a key driver of cell proliferation and survival in melanoma. This understanding has led to the development of targeted therapies such as vemurafenib and dabrafenib, which specifically inhibit the BRAFV600E mutation and have shown significant efficacy in treating patients harboring this genetic alteration.
In clinical practice, the detection of BRAF mutations is a critical factor in treatment decisions, particularly for metastatic melanoma. Testing for the BRAFV600 mutation is recommended for patients with distant metastases, non-resectable regional metastases, or high-risk stage III melanoma post-surgery [7]. Performing this test on metastatic tissue samples is ideal, due to the high concordance of BRAF mutation status in primary and metastatic lesions. The result of this testing significantly informs therapeutic decision-making, underscoring the role of genetic profiling in the personalized treatment of melanoma.
3.2. NRAS Mutations
NRAS mutations, which are found in 15–20% of melanoma cases, are significant drivers of the disease, affecting melanoma development through a distinct pathway [7]. These mutations activate the MAPK pathway, albeit through a mechanism different from that of BRAF mutations. NRAS mutations are typically mutually exclusive of BRAF mutations, reinforcing their unique role in melanoma’s molecular pathology [7]. The identification of NRAS mutations is not only essential for understanding melanoma’s genetic diversity, but also plays an important role in clinical decision-making, particularly in cases without BRAF mutations. Despite not being direct targets of current therapies, the presence of NRAS mutations guides the selection and tailoring of treatment strategies. So far, targeted therapies specifically addressing NRAS-mutated melanoma have shown limited success [5]. However, this area remains a significant focus of ongoing research, especially in the context of advanced melanoma that has not responded to standard immunotherapies, including anti-CTLA4 and anti-PD1 antibodies. The development of more effective treatment options for NRAS-mutated melanoma is a key objective of current clinical investigations, reflecting the continuous effort to improve therapeutic outcomes for this challenging subset of melanoma patients.
3.3. c-KIT Mutations
c-KIT mutations, while less common than BRAF and NRAS mutations, play a significant role in certain melanoma subtypes, particularly mucosal and acral melanomas. These mutations are predominantly found in melanomas arising from mucosal, acral, and chronically sun-damaged skin, representing a distinct subset within the broader spectrum of melanoma [7].
In the clinical setting, it is recommended to initially test for BRAF and NRAS mutations in acral and mucosal melanomas [5]. If those tests return negative, a further analysis for c-KIT mutations is advised [5]. This stepwise approach to genetic testing ensures a precise and targeted strategy for managing these specific melanoma subtypes.
Therapies targeting c-KIT mutations, although not yet formally approved, have shown promising results in treating melanomas harboring these genetic alterations. Clinical benefits from c-KIT inhibitors have been observed in selected patients, underscoring the importance of these mutations in the therapeutic landscape of melanoma [7]. The ongoing research and development of treatments targeting c-KIT mutations are pivotal in enhancing care and outcomes for patients with these specific melanoma subtypes.
3.4. GNAQ/GNA11 Mutations
GNAQ/GNA11 mutations, commonly identified in uveal melanomas [8], represent a distinct genetic subgroup within melanoma. Despite the current limitations of available treatments, these mutations are under active study for targeted therapy options, reflecting a growing interest in developing specific treatments for these subtypes.
In non-uveal melanomas, GNAQ/GNA11 mutations display a unique genetic profile characterized by a lower tumor mutational burden and fewer UV signature mutations than are common in cutaneous melanomas [9]. This suggests significant differences in the genetic landscape of these tumors compared with both cutaneous and uveal melanomas. Additionally, non-uveal melanomas with GNAQ/GNA11 mutations tend to metastasize lymphatically, similar to cutaneous melanoma, rather than the hematogenous metastasis typically seen in uveal melanoma [9]. These findings underline the urgent need for novel therapeutic approaches because non-uveal melanomas with GNAQ/GNA11 mutations respond poorly to existing systemic therapies, including immune checkpoint inhibitors (ICIs) [9]. The rarity and distinct behavior of GNAQ/GNA11 mutant non-uveal melanomas highlight the importance of ongoing research to develop effective treatments for this unique melanoma subgroup.
4. Molecular Pathways
4.1. MAPK/ERK Pathway
The MAPK/ERK pathway is central to melanoma, with alterations in this pathway often driving tumorigenesis [7]. This pathway, which includes RAS, RAF, ERK, and mitogen-activated extracellular signal-regulated kinase (MEK), is crucial for regulating cellular proliferation [1]. The discovery of activating NRAS mutations in melanoma in the mid-1980s and the subsequent identification of BRAF mutations in 2002 have been significant milestones in understanding melanoma’s molecular pathology [10]. Both of these mutations lead to the overactivation of the MAPK/ERK pathway, promoting melanoma cell proliferation and survival.
The exploration of the MAP kinase pathway as a therapeutic target began with responses observed from MEK inhibitors. However, the development of potent and selective inhibitors such as vemurafenib, dabrafenib, and encorafenib, which target mutated BRAF, has been the major breakthrough in melanoma treatment [10]. The late 2000s saw a pivotal shift with the introduction of these targeted therapies. The initial use of drugs such as sorafenib paved the way for more effective and selective BRAF inhibitors [10].
Combination therapies of BRAF/MEK inhibitors have significantly improved the efficacy of melanoma treatments, with reduced toxicity and longer progression-free survival (PFS) times compared with monotherapies [10]. These advances highlight the significance of the MAPK/ERK pathway in melanoma’s molecular pathology and response to therapy, underscoring the importance of understanding the influence of these mutations when developing effective treatments.
4.2. PI3K/AKT/mTOR Pathway
The PI3K/AKT/mTOR pathway plays a central role in melanoma development, affecting cell survival, proliferation, and metastasis. Its activation, often driven by genetic mutations and signaling imbalances, underscores the need for targeted therapeutic interventions. The constitutive activation of this pathway is a hallmark of melanoma’s aggressiveness and contributes to resistance against standard treatments by influencing key cellular processes such as autophagic cell death and cell cycle regulation [11].
A range of targeted therapies focusing on inhibitors of PI3K, AKT, and mTOR is under investigation and showing promising results in clinical trials, both as individual treatments and in combination with other drugs, such as BRAF and MEK inhibitors [11]. The exploration of natural compounds, repurposed drugs, and novel synthetic molecules targeting this pathway, along with the emerging role of miRNA in modulating this pathway, presents new opportunities for treatment and underscores the pathway’s significance for the development of novel, effective, and personalized melanoma therapies [11].
5. Melanogenesis and Neuroendocrine Regulation in Melanoma Progression
Recent studies have highlighted the complex roles of melanin and melanogenesis in melanoma, revealing their protective effects against UV radiation and their potential to promote malignant transformation [12]. The synthesis of eumelanin and pheomelanin, influenced by environmental and hormonal factors, offers both defense and risks, with the instability of pheomelanin contributing to a mutagenic environment [13][14][15][16][17][18][19][20][21][22][23]. This duality affects melanoma’s development and response to treatments, with advanced melanomas showing a negative correlation between pigmentation and survival, indicating melanin’s dual impact [24][25][26][27]. Inhibiting melanogenesis could, therefore, enhance therapeutic outcomes, highlighting melanin’s intricate influence on melanoma behavior.
Moreover, melanoma’s ability to influence both local and systemic physiological responses through the secretion of neuroendocrine factors, including proopiomelanocortin (POMC) peptides, corticotropin-releasing hormone (CRH), and glucocorticoids, underscores its complex role in the body’s regulatory systems [28]. POMC peptides, including melanocyte-stimulating hormone (MSH), are immunosuppressive, and an increased expression of POMC peptides was noted during the progression of melanomas to advanced stages [29][30][31][32][33][34][35][36][37]. This capability of melanoma to manipulate the neuroendocrine and immune responses contributes to its survival and progression, altering homeostasis in favor of the tumor [28]. Such interactions necessitate the exploration of therapeutic strategies aimed at targeting these pathways, offering potential to improve treatment outcomes and patient prognosis.
References
- Lazaroff, J.; Bolotin, D. Targeted Therapy and Immunotherapy in Melanoma. Dermatol. Clin. 2023, 41, 65–77.
- Di Raimondo, C.; Lozzi, F.; Di Domenico, P.P.; Campione, E.; Bianchi, L. The Diagnosis and Management of Cutaneous Metastases from Melanoma. Int. J. Mol. Sci. 2023, 24, 14535.
- Seth, R.; Agarwala, S.S.; Messersmith, H.; Alluri, K.C.; Ascierto, P.A.; Atkins, M.B.; Bollin, K.; Chacon, M.; Davis, N.; Faries, M.B.; et al. Systemic Therapy for Melanoma: ASCO Guideline Update. J. Clin. Oncol. 2023, 41, 4794–4820.
- Chen, H.; Hou, K.; Yu, J.; Wang, L.; Chen, X. Nanoparticle-Based Combination Therapy for Melanoma. Front. Oncol. 2022, 12, 928797.
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320.
- Yang, T.T.; Yu, S.; Ke, C.K.; Cheng, S.T. The Genomic Landscape of Melanoma and Its Therapeutic Implications. Genes 2023, 14, 1021.
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255.
- Silva-Rodríguez, P.; Fernández-Díaz, D.; Bande, M.; Pardo, M.; Loidi, L.; Blanco-Teijeiro, M.J. GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers 2022, 14, 3066.
- Livingstone, E.; Zaremba, A.; Horn, S.; Ugurel, S.; Casalini, B.; Schlaak, M.; Hassel, J.C.; Herbst, R.; Utikal, J.S.; Weide, B.; et al. GNAQ and GNA11 mutant nonuveal melanoma: A subtype distinct from both cutaneous and uveal melanoma. Br. J. Dermatol. 2020, 183, 928–939.
- Flaherty, K.T. A twenty year perspective on melanoma therapy. Pigment. Cell Melanoma Res. 2023, 36, 563–575.
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803.
- Slominski, R.M.; Sarna, T.; Plonka, P.M.; Raman, C.; Brozyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496.
- Pawelek, J.M.; Lerner, A.B. 5, 6-Dihydroxyindole is a melanin precursor showing potent cytotoxicity. Nature 1978, 276, 627–628.
- Slominski, A.; PAUs, R.; Mihm, M. Inhibition of melanogenesis as an adjuvant strategy in the treatment of melanotic melanomas: Selective review and hypothesis. Anticancer. Res. 1998, 18, 3709–3715.
- Miranda, M.; Ligas, C.; Amicarelli, F.; D’Alessandro, E.; Brisdelli, F.; Zarivi, O.; Poma, A. Sister chromatid exchange (SCE) rates in human melanoma cells as an index of mutagenesis. Mutagenesis 1997, 12, 233–236.
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The toxicity of melanin precursors. J. Investig. Dermatol. 1978, 70, 113–116.
- Wick, M.M. Levodopa/dopamine analogs as inhibitors of DNA synthesis in human melanoma cells. J. Investig. Dermatol. 1989, 92, S329–S331.
- Wick, M.; Fitzgerald, G. Inhibition of reverse transcriptase by tyrosinase generated quinones related to levodopa and dopamine. Chem.-Biol. Interact. 1981, 38, 99–107.
- Kim, E.; Panzella, L.; Napolitano, A.; Payne, G.F. Redox activities of melanins investigated by electrochemical reverse engineering: Implications for their roles in oxidative stress. J. Investig. Dermatol. 2020, 140, 537–543.
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of eumelanin and pheomelanin and its pathophysiological implications. Photochem. Photobiol. 2018, 94, 409–420.
- Brash, D.E. UV-induced melanin chemiexcitation: A new mode of melanoma pathogenesis. Toxicol. Pathol. 2016, 44, 552–554.
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847.
- Lembo, S.; Di Caprio, R.; Micillo, R.; Balato, A.; Monfrecola, G.; Panzella, L.; Napolitano, A. Light-independent pro-inflammatory and pro-oxidant effects of purified human hair melanins on keratinocyte cell cultures. Exp. Dermatol. 2017, 26, 592–594.
- Brożyna, A.A.; Jóźwicki, W.; Carlson, J.A.; Slominski, A.T. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum. Pathol. 2013, 44, 2071–2074.
- Brozyna, A.; Jozwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853.
- Shields, C.L.; Kaliki, S.; Furuta, M.; Fulco, E.; Alarcon, C.; Shields, J.A. American joint committee on cancer classification of uveal melanoma (anatomic stage) predicts prognosis in 7731 patients: The 2013 zimmerman lecture. Ophthalmology 2015, 122, 1180–1186.
- Shields, C.; Kaliki, S.; Cohen, M.; Shields, P.; Furuta, M.; Shields, J. Prognosis of uveal melanoma based on race in 8100 patients: The 2015 Doyne Lecture. Eye 2015, 29, 1027–1035.
- Slominski, R.M.; Raman, C.; Chen, J.Y.; Slominski, A.T. How cancer hijacks the body’s homeostasis through the neuroendocrine system. Trends Neurosci. 2023, 46, 263–275.
- Böhm, M.; Luger, T.A.; Tobin, D.J.; García-Borrón, J.C. Melanocortin receptor ligands: New horizons for skin biology and clinical dermatology. J. Investig. Dermatol. 2006, 126, 1966–1975.
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020.
- Hedley, S.; Murray, A.; Sisley, K.; Ghanem, G.; Morandini, R.; Gawkrodger, D.; Mac Neil, S. α-Melanocyte stimulating hormone can reduce T-cell interaction with melanoma cells in vitro. Melanoma Res. 2000, 10, 323–330.
- Slominski, A.; Wortsman, J.; Mazurkiewicz, J.E.; Matsuoka, L.; Dietrich, J.; Lawrence, K.; Gorbani, A.; Paus, R. Detection of proopiomelanocortin-derived antigens in normal and pathologic human skin. J. Lab. Clin. Med. 1993, 122, 658–666.
- Nagahama, M.; Funasaka, Y.; Fernandez-Frez, M.; Ohashi, A.; Chakraborty, A.; Ueda, M.; Ichihashi, M. Immunoreactivity of α-melanocyte-stimulating hormone, adrenocorticotrophic hormone and β-endorphin in cutaneous malignant melanoma and benign melanocytic naevi. Br. J. Dermatol. 1998, 138, 981–985.
- Ghanem, G.; Lienard, D.; Hanson, P.; Lejeune, F.; Fruhling, J. Increased serum alpha-melanocyte stimulating hormone (alpha-MSH) in human malignant melanoma. Eur. J. Cancer Clin. Oncol. 1986, 22, 535–536.
- Loir, B.; Bouchard, B.; Morandini, R.; Marmol, V.D.; Deraemaecker, R.; Garcia-Borron, J.C.; Ghanem, G. Immunoreactive α-melanotropin as an autocrine effector in human melanoma cells. Eur. J. Biochem. 1997, 244, 923–930.
- Funasaka, Y.; Sato, H.; Chakraborty, A.K.; Ohashi, A.; Chrousos, G.P.; Ichihashi, M. Expression of proopiomelanocortin, corticotropin-releasing hormone (CRH), and CRH receptor in melanoma cells, nevus cells, and normal human melanocytes. J. Investig. Dermatol. Symp. Proc. 1999, 4, 105–109.
- Sato, H.; Nagashima, Y.; Chrousos, G.P.; Ichihashi, M.; Funasaka, Y. The expression of corticotropin-releasing hormone in melanoma. Pigment Cell Res. 2002, 15, 98–103.
More
Information
Subjects:
Dermatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
463
Revisions:
2 times
(View History)
Update Date:
14 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No