Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aldona Kasprzak | -- | 4559 | 2024-03-12 12:10:55 | | | |
| 2 | Rita Xu | Meta information modification | 4559 | 2024-03-13 02:26:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kasprzak, A.; Geltz, A. Somatostatin in Colorectal Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/56160 (accessed on 25 June 2026).
Kasprzak A, Geltz A. Somatostatin in Colorectal Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/56160. Accessed June 25, 2026.
Kasprzak, Aldona, Agnieszka Geltz. "Somatostatin in Colorectal Cancer" Encyclopedia, https://encyclopedia.pub/entry/56160 (accessed June 25, 2026).
Kasprzak, A., & Geltz, A. (2024, March 12). Somatostatin in Colorectal Cancer. In Encyclopedia. https://encyclopedia.pub/entry/56160
Kasprzak, Aldona and Agnieszka Geltz. "Somatostatin in Colorectal Cancer." Encyclopedia. Web. 12 March, 2024.
Copy Citation
Somatostatin, a somatotropin release inhibiting factor (SST, SRIF), is a widely distributed multifunctional cyclic peptide and acts through a transmembrane G protein-coupled receptor (SST1-SST5). Over the past decades, research has begun to reveal the molecular mechanisms underlying the anticancer activity of this hormonal peptide.
neuroendocrine and non-endocrine tumors
colorectal cancer (CRC)
somatostatin (SST, SRIF)
1. Introduction
Colorectal cancer (CRC), which encompasses the colorectum (including the anus), referred to as ICD-10 C18-C21 [1], is the third most prevalent malignant tumor around the world (including Europe) and second among cancer-related death causes [2][3][4]. In Poland in 2020, in terms of incidence, it also ranked third among both sexes. As a cause of mortality, it was in second place in men and third in women [3]. About 90% of CRC cases are sporadic, caused by somatic mutations leading to invasive cancer [5].
Antitumor effects include primarily inhibiting cell proliferation and increasing cell apoptosis. Other processes (often associated with antiproliferative effects) include inhibition of angiogenesis and regulation of the tumor’s immune status [6][7]. In the case of CRC, potential antitumor effects also include eliminating chronic inflammatory changes, modifying the intestinal microbiome and regulating the intestinal barrier and the interaction between cancer and the tumor microenvironment (TME) [8][9][10][11][12]. Somatostatin, a somatotropin release inhibiting factor (SST, SRIF), discovered in the rat/ovine hypothalamus [13][14], is a widely distributed multifunctional cyclic peptide produced by cells scattered throughout the human body [15][16]. SST as an endogenous peptide hormone and its synthetic analogues (SSAs) acts through five types of SST receptors (SSTRs): SST1, SST2 (SST2A and SST2B in rodents) [17][18], SST3, SST4 and SST5, which belong to the superfamily of transmembrane G protein-coupled receptors (GPCR family). Functionally, SST and SSAs are related to well-known signal transduction pathways presented in other reviews [15][19][20][21][22][23].
The most commonly described effects of somatostatin receptor (SST1-5) activation by ligand (SST) are: (i) adenylyl cyclase (ACL) inhibition; (ii) activation of protein phosphotyrosine phosphatases (PTPs); and (iii) modulation of mitogen-activated protein kinase/extracellular signal regulated kinase (MAPK/ERK). Other antitumor effectors via SSTRs include (iv) phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT) and (v) calcium signaling pathways [21][22][23]. ACL inhibition leads to a decrease in adenosine monophosphate (cAMP), which results in the downregulation of protein kinase activity, which suppresses the activity of oncogenes and the development of cancer. Activation of PTPs leads to dephosphorylation and inactivation of tyrosine kinase [24]. Among the protein kinases inhibited by PTPs is MAPK, which results in inhibition of DNA and protein synthesis. This signaling pathway is also responsible for pro-apoptotic effects. In the PI3K/AKT pathway, there is an increase in the expression of p21, p27 and the tumor suppressor gene Zac1, which causes the accumulation of cells in the G1 phase without entering the S phase and the inhibition of cell proliferation. ACL inhibition is responsible for the antisecretory effect of SST/SSAs, as well as lowering the intracellular calcium concentration due to the inhibition of voltage-dependent Ca2+ channels and activation of K+ channels. These activities may also lead indirectly to the inhibition of proliferation [15][16][21][22][25]. The molecular mechanisms of the antitumor effects of SST have been described in various solid tumors [23][26][27][28][29].
Concerning gastrointestinal tract (GIT) tumors, the antitumor activity of SST has been well documented in gastroenteropancreatic neuroendocrine tumors (GEP-NETs) [23][30][31] and poorly in non-endocrine cancers, including sporadic CRC. There are many unresolved issues in research on the biological effects of SST in CRC. It is unclear, among others, the role of SST in CRC cell histogenesis [32][33][34][35][36] and in increasing the population of cancer stem cells (CSCs) [37][38]. Although SST was among the nine hub genes associated with the diagnosis and prognosis of CRC [39], its diagnostic and prognostic role in CRC in everyday clinical practice is poorly defined. The clinical value of examining circadian serum SST levels in CRC is also uncertain [40][41].
SSAs treatment mainly concerns highly differentiated neuroendocrine neoplasms (NENs) of GIT and other tumors that express SSTRs. Although the importance of SSAs in the treatment of symptomatic hormonally active tumors is widely recognized, their role as anticancer drugs is controversial and still undefined [42][43][44][45][46].
2. Antitumor Effects of Somatostatin in CRC
There are direct and indirect antitumor effects of SST and SSAs in various cancers. The direct action occurs through SSTRs present on cancer cells and includes inhibition of mitogenic signals dependent on growth factors, induction of apoptosis and inhibition of cell cycle and/or tumor growth. The indirect effect consists of inhibiting the exocrine and endocrine secretion of growth factors or trophic hormones, e.g., epidermal growth factor (EGF), basic fibroblast growth factor (bFGF) and/or insulin-like growth factor 1/2 (IGF-1/2). Indirect actions also include immune modulating effects and inhibition of tumor angiogenesis [23][26][27][28][47][48].
2.1. Inhibition of Cell Proliferation
Depending on the SSTR subtype, different signal transduction pathways are involved in the antiproliferative effect of SST, which have been studied in more detail using various in vivo and in vitro research models [19][26].
2.1.1. Evidence from In Vivo Studies
Higher circadian SST concentrations were observed both in ulcerative colitis (UC) [49], in patients with colorectal polyps and in CRC compared to healthy people. This suggests a protective effect of SST in precancerous alterations of the colon and CRC [40]. Other studies by these authors conducted on CRC patients do not seem to confirm the antitrophic effect of SST at the cellular and subcellular levels [41].
Many studies have demonstrated the tissue expression of SST (mRNA, protein) and characterized the cells producing SST in this type of tumor. Most studies localize this peptide both in EECs, where colocalization with other NPs is common, e.g., chromogranin A, serotonin, glucagon, bombesin, vasoactive intestinal peptide (VIP), and in cancer cells (reviewed in [50]). Amphocrine features of CRC cells have been demonstrated [51][52]. This may confirm the theory of the origin of EECs [34] or the development of this type of tumor from multipotential endodermal SCs [32][34]. Based on in vivo and in vitro studies, it has been shown that SST signaling controls the rate of NCs maturation as SCs mature along the NE cell lineage, which contributes to SC silencing and inhibition of cell proliferation [37]. Additionally, SST expression was demonstrated in the structures of the nervous system present in the tumor, where it also co-expressed with other NPs, e.g., protein gene product 9.5 (PGP 9.5), substance P (SP) and calcitonin gene-related peptide (CGRP) [53].
Typically, decreased SST expression was observed in CRC compared to normal colon tissues [54][55][56][57] or was not detected at all either at the protein [58] or mRNA [37] level. This was confirmed by studies on an animal model (male rats with 1,2-dimethylhydrazine dihydrochloride-induced colonic adenocarcinoma), which showed only a few SST-positive cells [59]. Low SST tissue expression suggests loss of the inhibitory role of SST in tumor growth. Other authors consider the decrease in SST expression and the increase in ectopic expression of other NPs as indicators of preneoplastic changes in the large intestine [56]. A reduction in cellular SST expression in CRC is correlated with poor grading and staging [55]. However, an increase in the number of highly differentiated NCs compared to the number of poorly differentiated NCs in CRC was also observed [60]. The latest bioinformatic analysis indicates six hub genes, including SST and SST2, that were significantly downregulated in colon adenocarcinoma compared to controls [61]. SST deficiency or abnormal function of its receptors would be risk factors for the development of CRC.
Administration of various SSAs resulted in a reduction in tissue expression of proliferative antigens, i.e., Ki-67 in 4/12 patients with rectal cancer (RC) [62] and proliferating cell nuclear antigen (PCNA) in 6/10 patients with CRC versus control [63]. Moreover, SSAs treatment reduced serum levels of GH and IGF-1 [64][65][66][67]. A decrease in the average percentage of cells in the S phase was also observed due to the simultaneous reduction in IGF-1 in the blood serum of these patients, but without changes in GH and EGF concentrations [67]. Another study using lanreotide (LAN) in advanced CRC did not confirm the antitumor activity of this SSA. Only higher doses of LAN seemed to maintain reduced IGF-1 concentrations in the blood serum of these patients [66].
The mechanisms of the antiproliferative effect of natural SST in CRC can be also associated with the abnormal expression of cyclins (D1, A, E) and cyclin-dependent kinases (CDK2 and CDK4) in CRC tissues. SST would regulate the stage of entry into the S phase of the cell cycle [68].
2.1.2. Evidence from In Vitro and Animal Model Studies
Inhibition of proliferation, increase in apoptosis of CRC cells, inhibition of tumor growth in animals (nude mice bearing xenografts) and reduction in angiogenesis after the use of SSAs have been proven in numerous studies both in vitro and in vivo. The most common CRC cell lines tested for this purpose were, in descending order, HT-29, HCT-116, Caco-2, SW480 and SW620. All these lines are formed by cells with an epithelial phenotype derived from patients with colorectal adenocarcinoma. The most frequently represented work is performed on the HT-29 cell line which is derived from a human colon adenocarcinoma originating from a 44-year-old female [69]. HT-29 cells showed particularly high susceptibility to several modified octapeptide analogues of SST containing unnatural amino acids (AA) compared to other cell lines (MDA-MB-23, HepG2, HeLa and Lep-3). The most pronounced antiproliferative effects were demonstrated by the compound 4 C: Orn5 and α-aminoisobutyric acid (6) (Aib6) in these cells with the IC50 = 0.0199 μM [70].
HCT-116 and HT-29 represent CRC cell lines that correspond to the more (Dukes’ D) and less aggressive forms (Dukes’ C) of CRC, respectively [71][72]. HCT-116 cells were originally isolated from primary tumors derived from colon ascendens of a 48-year-old male [73]. Cells from these lines possessed different statuses of one of the most commonly mutated genes in CRC, i.e., KRAS. HT-29 cells which have microsatellite stable (MSS) status had wild-type (wt) KRAS and PTEN, but BRAF, PIK3CA and TP53 mutations (R273H), whereas HCT-116 cells, which have MSI status, gained mutated KRAS and PIK3CA [71].
In HT-29 cells and nude mouse xenografts, an obvious inhibition of proliferation was demonstrated after SST/SSA treatment [70][74][75][76][77][78][79][80][81]. Some authors observed the antiproliferative effect of SST, but only in the presence of serum [75] or in a time and dose-dependent manner [70][79][80][81]. Differences in the intensity of the antiproliferative effect of two SSAs (Sandostatin and TT-232) on HT-29 cells were demonstrated. After treatment with TT-232, a 59 ± 6% decrease in the cancer cell number was observed, and after Sandostatin, only 21 ± 12% [77]. However, Keri et al. showed that TT-232 was effective in inhibiting tumor growth (up to 70% inhibition) in the case of transplanted animal tumors (including C26) and human tumor xenografts. In the case of HT-29 cells, the inhibition of proliferation also reached 72 ± 5% [82].
The antiproliferative effect of SSAs has also been described in other CRC cell lines, e.g., CX1 [74], LIM 1215, LIM 1863, LIM 2405, LIM 2412 [83], SW480 [84], SW620 [82][85], HCT-116 [86] and Caco-2 [80]. It was not observed in individual cases of the CRC cell lines tested, e.g., X56 [74], and rat colon cancer cells DHD/K12 [87]. Only a minimal effect was observed after administration of Sandostatin in the case of C170 and LIM 1215 cells [88]. No effect of SST on the invasive potential of murine colon adenocarcinoma cells 26L5 was observed [89].
OCT dose-dependent inhibition of proliferation and cell arrest in the G1 phase of the cell cycle were also observed in SW480 cells [90]. Szepeshazi et al. showed that another SST derivative, AN-238, inhibits the growth of experimental colon cancers (HT-29 and HCT-15 cells) that express SSTRs, regardless of their p53 status [91]. No significant differences in the intensity of cell proliferation were observed in mice injected with human colon cancer cells using triple therapy with OCT + galanin + serotonin versus 5-fluorouracil/leucovorin (5-FU/LV) [92] or compared to 5-FU/LV-irinotecan or 5FU/LV-oxaliplatin [93].
To further assess the direct antiproliferative mechanisms of SST, the presence of SSTRs (or specific binding sites) on cultured CRC cells was also examined as a condition for the action of SST/SSAs. In the case of HT-29 cells, binding sites for SST, bombesin and EGF [76], the existence of low-affinity SSTRs in such cells [94], high-affinity binding sites for SST [86] and functional SSTRs was shown [91]. Using the immunofluorescence technique (IF), the presence of receptor subtypes such as SST3/4/5 [95] and all types of SSTRs, including two isoforms of SST2 (SST2A and SST2B), was demonstrated. The presence of SST1/2/5 in HT-29 cells was demonstrated using RT-PCR [80]. In the case of Caco-2 cells, the presence of SST3/5 was demonstrated and in HCT-116 cells, SST2/3/5 [95].
Research by some authors showed that SST preferred SST3 and SST5 in their effects [81][95]. However, no functional SSTRs were detected on LoVo cells; hence, none of the SSAs used inhibited the proliferation of LoVo tumors [91]. LoVo cells are cells from a 56-year-old male, with Dukes’ C, with MSI status, with a KRAS mutation, but without mutations in other important genes of intestinal carcinogenesis (e.g., BRAF, PIK3CA, PTEN, TP53) [71].
It seems that greater antiproliferative effects occur when SSAs are used in combination with other antitumor agents. The use of a combination of OCT with interleukin 2 (IL-2) and interferon gamma (IFN-γ) gave a stronger antiproliferative effect on the growth of SW620 cells but not in SW480 cells [84]. Similarly, the use of Sandostatin together with 5-FU gave a stronger antiproliferative effect than the administration of Sandostatin alone in C170 and LIM 1215 cells [88]. These observations are confirmed by the study by Massari et al., who in the colon cancer cell line WiDr (identical with HT-29 cells), expressing a mutant p53 (mp53), showed that the SMS analogue has pro-apoptotic and antiproliferative effects, which can enhance the effect of 5-FU on human CRC cells expressing mp53 [96].
Recent studies also indicate better antiproliferative effects after using conjugated cetuximab (CTX)–OCT loaded onto Ca–alginate beads (CTX-OCT-Alg) compared to free drug. The studies were performed on three different cell lines, including CRC cells (HCT-116) [97]. The latest work by Fan et al. demonstrates a combined antitumor effect on cells with the presence of SSTRs, as well as a mouse model treated with thymidine kinase (TK) deleted vaccinia virus Tian Tan strain Guang9 (VG9/TK-) or VG9/(SST-14)2-human serum albumin (HSA). Fusion technology was used to extend the half-life of SST in the circulation by combining natural SST with a full-length HSA molecule. It was shown that VG9/(SST-14)2-HSA is more effective in prolonging the survival of all mice in both groups than VG9/TK-. However, the oncolytic activity of vaccinia viruses was not high enough in some cells, including HCT-116 cells, indicating that these cells were more resistant to the effects of the viruses. However, as a whole, these studies indicate that vaccinia VG9/(SST-14)2-HSA has oncolytic activity of the virus as well as anticancer activity [98].
2.1.3. Mechanisms of the Antiproliferative Action of SST
Studies on the mechanisms of action of SST/SSAs in CRC cell lines or animal models confirm the involvement of known intracellular pathways that regulate secretory activity, inhibit cell proliferation and increase cell apoptosis [15][16][20][22][47][48].
The antiproliferative mechanisms of SST/SSAs in CRC are related to the regulation of protein phosphorylation on tyrosine residues, which is an important cell signaling mechanism [16]. The control of protein phosphorylation/dephosphorylation occurs through the combined actions of protein-tyrosine kinases (PTKs) and protein-tyrosine phosphatases (PTPs), respectively [99]. They are essential for cellular homeostasis and can lead to disruptions in various important cellular pathways, including cell proliferation and differentiation. It is also known that >80% of all oncogenes encode PTKs, and PTPs that can reverse the action of PTKs play an important role as tumor suppressors [24].
As research by Keri et al. showed, a structural derivative of SST, with a five-residue ring (D-Phe-Cys-Tyr-D-Trp-Lys-Cys-Thr-NH2) called TT-232 inhibited the tyrosine kinase activity of some human carcinomas cell lines of the colon, and this inhibition correlated well with the antiproliferative effect but did not correlate with GH release inhibition [100][101]. Subsequent studies by these authors confirmed strong inhibition of tyrosine kinase activity (75%) after long-term incubation (24 h) with TT-232 in SW620 cell culture. Moreover, they showed that this effect correlated well with the inhibition of proliferation and the effect of inducing cell apoptosis. In addition, this study demonstrated the antiproliferative effect of TT-232 also on Colo205 cells (proliferation inhibition > 50%) and in an animal model (tumor growth inhibition ~70%) [82]. Other studies by the same group confirmed previous observations (strong antiproliferative effect of SW620 cells after TT-232) and additionally showed a rapid and sustained (5–30 min) increase in PTP activity [85]. It should be added that there are approximately 100 PTPs in the genome, approximately equivalent to the number of tyrosine kinases [102]. TT-232-induced PTP activation may therefore be an important early step in the signaling pathway in inhibiting cell proliferation in CRC [85]. Modulation of the activity of various PTPs is one of the intracellular pathways responsible for inhibiting cell growth also by OCT [80]. SST-stimulated PTP activity shares biochemical features with SHP1 and SHP2 phosphatases. These phosphatases belong to a family of cytosolic PTPs that contain motifs called src homology 2 (SH2) domains and are involved in protein–protein interactions through their association with specific phosphotyrosine residues [103]. The PTP family also includes density-enhanced phosphatase-1 (DEP-1) (humans)/PTPη (in rats). All are intracellular effectors of SSTRs [16].
Another mechanism of the antiproliferative effect of SST (also involving PTPs) in CRC cells (Caco-2, HT-29 and HCT-116) is the inhibition of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2). In Caco-2 cells, SST-14 has been shown to inhibit basal COX-2 expression, PGE2 production, DNA synthesis and cell growth. The inhibitory effect of COX-2 expression and function occurs through the activation of SST3 or SST5. Therefore, SST may oppose proliferative stimuli by reducing COX-2 expression driven by a negative regulation of protein kinase C-dependent mitogen-activated protein kinase (MAPK) and AKT activation. The attenuation of constitutive COX-2 expression by SST in CRC cells via SST3/5 is expected to occur through activation of PTP, which leads to the inhibition of MAPK signaling and is the main mechanism for inhibiting the growth of CRC cells [95].
Uncontrolled growth of CRC cells is also enabled by the activation of human telomerase reverse transcriptase (hTERT), a catalytic component of the telomerase complex. Telomerase, which maintains telomere length and maintains the cell’s replicative potential, is activated during the adenoma–carcinoma sequence and its activity increases during tumor progression. It is believed that telomere shortening plays a role in the early stages of colorectal carcinogenesis, resulting in chromosome instability [104]. The telomerase signaling was also examined in HT-29 and Caco-2 cells after OCT treatment. Increased telomerase activity in HT-29 cells cultured in the absence of serum and in the presence of 10% fetal bovine serum (FBS) was demonstrated. However, in Caco-2 cells, a decrease in telomerase activity was observed in cells cultured without serum and an increase in the presence of TBS. The authors speculate that OCT may inhibit cell proliferation selectively in Caco-2 cells by reducing telomerase activity, whereas in HT-29 cells it appears to inhibit cell proliferation through different molecular pathways [80].
An important mechanism of SST action is also the regulation of the distribution and expression of p86 Ku protein (Ku86), the regulatory subunit of DNA-dependent kinase and the SST binding site. Ku86 has been shown to behave as a specific nuclear receptor for SST and regulate p53 expression and apoptosis [82][105][106]. Quite early, it was shown that Ku86 modulates in vitro dephosphorylation of p34CDK2–phosphorylated histone H1 by phosphoprotein phosphatase 2A (PP2A) [105]. PP2A is one of the phosphoprotein phosphatases (PPPs), the largest family of phosphatases, and dephosphorylates hundreds of substrates involved in the cell cycle, regulating almost all major pathways (including MAPK and Wnt/β–catenin pathways) and cell cycle checkpoints [102]. These findings suggest that Ku86, as a nuclear SST receptor, may mediate negative control of cell cycle regulation by SST [105]. This is confirmed by other studies [77][106]. In addition to the quantitatively differentiated antiproliferative effect of Sandostatin (smaller) and TT-232 (larger) in HT-29 cells, the involvement of SST in the translocation of Ku86 from the cytosol to the cell nucleus was demonstrated [77]. However, studies with SST treatment of Caco-2 cells showed inhibition of cell growth while modulating the activation of the Ku70/86 heterodimer by SST. After SST treatment, an increase in Ku86 mRNA levels was observed in the cell nucleus. These studies confirm the hypothesis that SST controls cell cycle progression and DNA repair through a signaling pathway involving the regulation of Ku86 levels and Ku70/86 activity in the cell nucleus [106]. Further studies on Caco-2 cells showed that SST increases the binding between Ku70 and Ku86, induces an antiproliferative effect after 24 h and restores apoptosis in cells [107]. In turn, a new mechanism of cellular adaptation as a defense system against severe genomic stress caused by the functional loss of Ku70 has recently been discovered. Conditional deletion of XRCC6 (the gene encoding Ku70) has been shown to promote an adaptive, opportunistic transition to a parasitic lifestyle of HCT-116 cells at the expense of continuous host cell exploitation [108].
In the SW480 cells model, it was shown that OCT can inhibit the growth of human colonic cancer cells also by inhibiting the Wnt/β–catenin signaling pathway [90]. They also showed correlations of reduced tumor growth with clinical tumor biomarkers after the use of SSAs. A 13% decrease in carcinoembryonic antigen (CEA) level was observed in LoVo cells after treatment with SMS 201.995. Similarly, in xenografts, a correlation was detected between a decrease in tumor growth and a reduced serum level of CEA. The CEA concentration therefore reflected the number of cells in vitro and the size of the tumor in vivo in response to treatment with this type of SSA [109].
2.2. Pro-Apoptotic Effects of Somatostatin
2.2.1. Evidence from In Vivo Studies
There are few studies on the influence of SST on the apoptosis process in CRC in vivo. Mao et al. showed higher tissue expression of the pro-apoptotic protein Bax and lower expression of the anti-apoptotic protein Bcl-2 in CRC groups with high and moderate SST expression compared to the low expression group [110]. Similarly, apoptosis rates determined by the expression of Fas protein and two caspases (3 and 8) were higher in the groups of CRC patients with high and moderate SST expression compared to the low expression group [111].
2.2.2. Evidence from In Vitro and Animal Model Studies
The pro-apoptotic effects of SST/SSAs in vitro or in animal models were most often demonstrated in parallel with their antiproliferative effect or tumor growth inhibitory effect [57][82][86][90][96][112][113]. In HT-29 cells, after using TT-232, a 7-fold increase in the number of cells in apoptosis was demonstrated. However, in SW620 cells, a positive correlation was obtained between the increase in apoptosis and the antiproliferative effect in long-term incubation (24 h) with this SSA. The studies were confirmed in a mouse model, where a 70% inhibition of tumor growth was achieved in the transplanted Colon 26 tumor. The apoptosis-inducing effect was independent of p53, as there was no significant effect of TT-232 on the translocation of Ku86 from the cytosol to the cell nucleus [82]. However, in the murine transplantable Colon 38 cancer model, it was shown that when administered separately, both SMS and melatonin (Mel) significantly reduced the index of cell proliferation (labeling index, LI) and increased the apoptotic index (AI). However, no additive effect of SMS and Mel on cell proliferation or apoptosis was observed. The imbalance between the processes of proliferation and apoptosis has changed in favor of cell death [112]. Massari et al. showed the pro-apoptotic activity of the SMS 201.995, which was demonstrated together with the inhibition of cell proliferation in the WiDr cell line (identical to HT-29) [96]. Hohla et al. showed an increase in the number of apoptotic cells and inhibition of the proliferation of HTC-116 and P388/R84 cells in the S/G2 phase [86]. Inhibition of tumor growth and induction of apoptosis also depend on the OCT dose, as demonstrated in SW480 cells [90]. The same cells showed an increase in apoptosis mediated by SST2 and SST5 [113]. However, the administration of OCT to Caco-2 cell cultures increased in the percentage of cells with apoptosis, increased DNA fragmentation (Sub-G1 population) and decreased proliferation [57].
El-Salhy et al. in their work using a mouse model with implanted rat or human colonic adenocarcinoma cells and the administration of triple therapy (OCT + galanin + serotonin) obtained an increase in apoptosis and a decrease in the number of blood vessels, without a decrease in the proliferative index (PI) compared to control [114]. When the effects of triple therapy (including OCT) were compared with LV/5-FU therapy, a decrease in PI and the number of blood vessels was demonstrated and an increase in AI in mice treated with both LV/FU-triple therapy and with triple therapy only as compared with LV/FU-treated mice [115]. Another two studies on xenografts confirmed an increase in AI and a decrease in the number of tumor blood vessels compared to the control after the use of triple therapy (including OCT) [116] and an additional decrease in PI [117]. The reduction in tumor volume and weight after triple therapy appeared to be due to low proliferation and increased apoptosis and decreased tumor vascularity [116]. In subsequent studies by these authors, no significant differences were observed in relation to cellular apoptosis in mice injected with human colon cancer cells after the use of 5-FU/LV [92], 5-FU/LV-irinotecan or 5FU/LV-oxaliplatin [118] compared with triple therapy.
Interesting research on the apoptosis process concerns the use of a combined preparation consisting of four NPs (including SST) called DRF 7295. This peptide caused an increase in p53 levels, downregulation of Bcl-2 levels in Colo205 cells and induction of active caspase-3 in HT-29 cells [119][120].
2.2.3. Mechanisms of the Pro-Apoptotic Action of SST
The study of the pro-apoptotic activities of SST/SSAs in CRC in vitro resulted in the demonstration of certain mechanisms of apoptosis induction in cancer cells. The work of Keri et al. demonstrated strong inhibition of tyrosine kinase activity (75%) after long-term incubation (24 h) with TT-232 in SW620 cells, which correlated with the apoptosis-inducing effect [82]. An important mechanism of action of SST in modulating apoptosis in Caco-2 cells was determined via the interaction between Ku70, a nuclear isoform of clusterin (nCLU) and Bax. Increased levels of nCLU and significant induction of Bax were observed after SST treatment. The 55 kD nCLU is important in the apoptosis process and acts as a chaperone in DNA repair by binding Ku70. A 10-fold increase in the interaction between Ku70 and nCLU was observed after SST treatment compared to untreated cells. However, after 24 h of SST treatment, Bax was released and nCLU and Ku70 colocalized in the cell membrane and nucleus [107].
Another mechanism of OCT inducing apoptosis, demonstrated in SW480 cells, might be the negative regulation of the Wnt/β–catenin signaling pathway via SST2 and SST5. After OCT treatment of cancer cells, accumulation of β-catenin in plasmalemma, inhibition of T-cell factor-dependent transcription, downregulated Wnt target genes (cyclin D1 and c-Myc) and mediation of activation of glycogen synthase kinase 3β (GSK-3β) [113] were observed.
A summary of the mechanisms of action of SST to inhibit CRC cell proliferation and induce cell apoptosis, as well as inhibiting angiogenesis using SSAs, is shown in Figure 1.
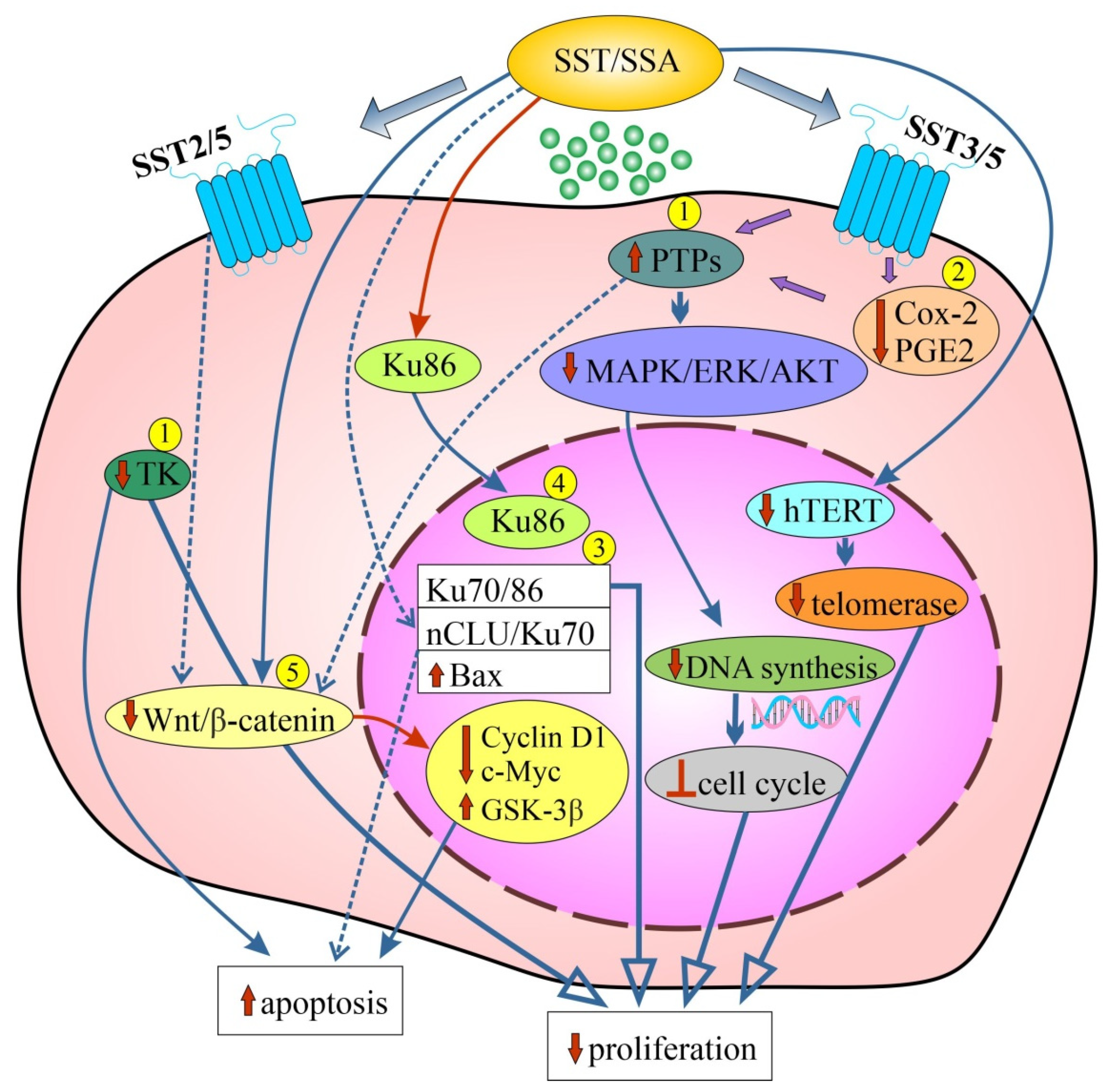
Figure 1. Schematic representation of the effects of SST related to inhibition of cell proliferation and stimulation of apoptosis in various colorectal cancer (CRC) cell lines (1–5) and/or in CRC animal models using SSAs (please refer to the main text for more details). Dashed lines with arrows indicate regulation/modulation of other signaling molecules/pathways. [↓/↑-reduced/increased expression/activity; Ʇ: inhibition, blockade; 1: SW620 cells; 2: Caco-2 cells; HT-29 cells; HCT-116 cells; 3: Caco-2 cells; 4: HT-29 cells; 5: SW480 cells; Cox: 2-cyclooxygenase-2; GSK: 3β-glycogen synthase kinase 3β; hTERT: human telomerase reverse transcriptase; MAPK/ERK/AKT: mitogen-activated protein kinase/extracellular signal regulated kinase/serine/threonine-protein kinase (protein kinase B); nCLU: nuclear clusterin; PGE2: prostaglandin E2; PTPs: protein-tyrosine phosphatases; SSA: synthetic somatostatin analogue; SST: somatostatin; SST2/3/5: somatostatin receptors 2/3/5; TK: tyrosine kinase; Wnt/β catenin: wingless + integrated or int-1/beta-catenin].
References
- World Health Organization. Tenth Revision. In International Statistical Classification of Diseases and Related Health Problems; WHO: Geneva, Switzerland, 2013; Volume 2.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer. 2021, 157, 308–347.
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344.
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532.
- Senga, S.S.; Grose, R.P. Hallmarks of cancer—The new testament. Open Biol. 2021, 11, 200358.
- Karin, M.; Shalapour, S. Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell Mol. Immunol. 2022, 19, 59–66.
- Park, C.H.; Eun, C.S.; Han, D.S. Intestinal microbiota, chronic inflammation, and colorectal cancer. Intest. Res. 2018, 16, 338–345.
- Li, Y.; Li, X.; Geng, C.; Guo, Y.; Wang, C. Somatostatin receptor 5 is critical for protecting intestinal barrier function in vivo and in vitro. Mol. Cell. Endocrinol. 2021, 535, 111390.
- Overacre-Delgoffe, A.E.; Bumgarner, H.J.; Cillo, A.R.; Burr, A.H.P.; Tometich, J.T.; Bhattacharjee, A.; Bruno, T.C.; Vignali, D.A.A.; Hand, T.W. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity 2021, 54, 2812–2824.e4.
- Schmitt, M.; Greten, F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667.
- Xie, Y.; Xie, F.; Zhou, X.; Zhang, L.; Yang, B.; Huang, J.; Wang, F.; Yan, H.; Zeng, L.; Zhang, L.; et al. Microbiota in Tumors: From Understanding to Application. Adv. Sci. 2022, 9, e2200470.
- Krulich, L.; Dhariwal, A.P.; McCann, S.M. Stimulatory and inhibitory effects of purified hypothalamic extracts on growth hormone release from rat pituitary in vitro. Endocrinology 1968, 83, 783–790.
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973, 179, 77–79.
- Patel, Y.C. Somatostatin and its receptor family. Front. Neuroendocrinol. 1999, 20, 157–198.
- Florio, T. Somatostatin/somatostatin receptor signalling: Phosphotyrosine phosphatases. Mol. Cell. Endocrinol. 2008, 286, 40–48.
- Vanetti, M.; Kouba, M.; Wang, X.; Vogt, G.; Höllt, V. Cloning and expression of a novel mouse somatostatin receptor (SSTR2B). FEBS Lett. 1992, 311, 290–294.
- Schindler, M.; Kidd, E.J.; Carruthers, A.M.; Wyatt, M.A.; Jarvie, E.M.; Sellers, L.A.; Feniuk, W.; Humphrey, P.P. Molecular cloning and functional characterization of a rat somatostatin sst2(b) receptor splice variant. Br. J. Pharmacol. 1998, 125, 209–217.
- Benali, N.; Ferjoux, G.; Puente, E.; Buscail, L.; Susini, C. Somatostatin receptors. Digestion 2000, 62 (Suppl. S1), 27–32.
- Lahlou, H.; Guillermet, J.; Hortala, M.; Vernejoul, F.; Pyronnet, S.; Bousquet, C.; Susini, C. Molecular signaling of somatostatin receptors. Ann. N. Y. Acad. Sci. 2004, 1014, 121–131.
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Würth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide receptor targeting in cancer: The somatostatin paradigm. Int. J. Pept. 2013, 2013, 926295.
- Gomes-Porras, M.; Cárdenas-Salas, J.; Álvarez-Escolá, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682.
- Kumar, U. Somatostatin and Somatostatin Receptors in Tumour Biology. Int. J. Mol. Sci. 2023, 25, 436.
- Laczmanska, I.; Sasiadek, M.M. Tyrosine phosphatases as a superfamily of tumor suppressors in colorectal cancer. Acta Biochim. Pol. 2011, 58, 467–470.
- Gatto, F.; Barbieri, F.; Arvigo, M.; Thellung, S.; Amarù, J.; Albertelli, M.; Ferone, D.; Florio, T. Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. Int. J. Mol. Sci. 2019, 20, 3940.
- Ferjoux, G.; Bousquet, C.; Cordelier, P.; Benali, N.; Lopez, F.; Rochaix, P.; Buscail, L.; Susini, C. Signal transduction of somatostatin receptors negatively controlling cell proliferation. J. Physiol. Paris 2000, 94, 205–210.
- Pyronnet, S.; Bousquet, C.; Najib, S.; Azar, R.; Laklai, H.; Susini, C. Antitumor effects of somatostatin. Mol. Cell. Endocrinol. 2008, 286, 230–237.
- Keskin, O.; Yalcin, S. A review of the use of somatostatin analogs in oncology. OncoTargets Ther. 2013, 6, 471–483.
- Ramírez-Perdomo, A.; Márquez-Barrios, G.; Gutiérrez-Castañeda, L.D.; Parra-Medina, R. Neuroendocrine Peptides in the pathogenesis of colorectal carcinoma. Exp. Oncol. 2023, 45, 3–16.
- Strosberg, J.; Kvols, L. Antiproliferative effect of somatostatin analogs in gastroenteropancreatic neuroendocrine tumors. World J. Gastroenterol. 2010, 16, 2963–2970.
- Baldelli, R.; Barnabei, A.; Rizza, L.; Isidori, A.M.; Rota, F.; Di Giacinto, P.; Paoloni, A.; Torino, F.; Corsello, S.M.; Lenzi, A.; et al. Somatostatin analogs therapy in gastroenteropancreatic neuroendocrine tumors: Current aspects and new perspectives. Front. Endocrinol. 2014, 5, 7.
- Ohmori, T.; Okada, K.; Tabei, R. Immunoreactivity to neurohormonal polypeptide in colorectal carcinomas and tumor-neighboring mucosa, and its significance. Arch. Pathol. Lab. Med. 1996, 120, 560–568.
- Ohmori, T.; Asahi, S.; Sato, C.; Maki, F.; Masumoto, A.; Okada, K. Bcl-2 protein expression and gut neurohormonal polypeptide/amine production in colorectal carcinomas and tumor-neighboring mucosa, which closely correlate to the occurrence of tumor. Histol. Histopathol. 1999, 14, 37–44.
- Swatek, J.; Chibowski, D. Endocrine cells in colorectal carcinomas. Immunohistochemical study. Pol. J. Pathol. 2000, 51, 127–136.
- Jesinghaus, M.; Konukiewitz, B.; Keller, G.; Kloor, M.; Steiger, K.; Reiche, M.; Penzel, R.; Endris, V.; Arsenic, R.; Hermann, G.; et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod. Pathol. 2017, 30, 610–619.
- Ogimi, T.; Sadahiro, S.; Kamei, Y.; Chan, L.F.; Miyakita, H.; Saito, G.; Okada, K.; Suzuki, T.; Kajiwara, H. Distribution of Neuroendocrine Marker-Positive Cells in Colorectal Cancer Tissue and Normal Mucosal Tissue: Consideration of Histogenesis of Neuroendocrine Cancer. Oncology 2019, 97, 294–300.
- Modarai, S.R.; Opdenaker, L.M.; Viswanathan, V.; Fields, J.Z.; Boman, B.M. Somatostatin signaling via SSTR1 contributes to the quiescence of colon cancer stem cells. BMC Cancer 2016, 16, 941.
- Zhang, T.; Ahn, K.; Emerick, B.; Modarai, S.R.; Opdenaker, L.M.; Palazzo, J.; Schleiniger, G.; Fields, J.Z.; Boman, B.M. APC mutations in human colon lead to decreased neuroendocrine maturation of ALDH+ stem cells that alters GLP-2 and SST feedback signaling: Clue to a link between WNT and retinoic acid signalling in colon cancer development. PLoS ONE 2020, 15, e0239601.
- Chen, L.; Lu, D.; Sun, K.; Xu, Y.; Hu, P.; Li, X.; Xu, F. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene 2019, 692, 119–125.
- Payer, J., Jr.; Huorka, M.; Duris, I.; Mikulecky, M.; Kratochvilovà, H.; Ondrejka, P. Somatostatin and large bowel polyps. Hepatogastroenterology 1995, 42, 775–777.
- Payer, J., Jr.; Huorka, M.; Duris, I.; Ondrejka, P.; Kratochvílova, H.; Ilkova, M.; Holéczy, P. Circadian rhythmicity of plasma somatostatin, gastrin and cortisol in colon cancer patients. Hepatogastroenterology 1997, 44, 72–77.
- Hejna, M.; Schmidinger, M.; Raderer, M. The clinical role of somatostatin analogues as antineoplastic agents: Much ado about nothing? Ann. Oncol. 2002, 13, 653–668.
- Rai, U.; Thrimawithana, T.R.; Valery, C.; Young, S.A. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol. Ther. 2015, 152, 98–110.
- Diamantopoulos, L.N.; Laskaratos, F.M.; Kalligeros, M.; Shah, R.; Navalkissoor, S.; Gnanasegaran, G.; Banks, J.; Smith, J.; Jacobs, B.; Galanopoulos, M.; et al. Antiproliferative Effect of Above-Label Doses of Somatostatin Analogs for the Management of Gastroenteropancreatic Neuroendocrine Tumors. Neuroendocrinology 2021, 111, 650–659.
- Rinzivillo, M.; De Felice, I.; Magi, L.; Annibale, B.; Panzuto, F. Octreotide long-acting release (LAR) in combination with other therapies for treatment of neuroendocrine neoplasia: A systematic review. J. Gastrointest. Oncol. 2021, 12, 845–855.
- Hu, Y.; Ye, Z.; Wang, F.; Qin, Y.; Xu, X.; Yu, X.; Ji, S. Role of Somatostatin Receptor in Pancreatic Neuroendocrine Tumor Development, Diagnosis, and Therapy. Front. Endocrinol. 2021, 12, 679000.
- Susini, C.; Buscail, L. Rationale for the use of somatostatin analogs as antitumor agents. Ann. Oncol. 2006, 17, 1733–1742.
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252.
- Payer, J.; Huorka, M.; Duris, I.; Mikulecky, M.; Kratochvílová, H.; Ondrejka, P.; Lukác, L. Plasma somatostatin levels in ulcerative colitis. Hepatogastroenterology 1994, 41, 552–553.
- Kasprzak, A. Somatostatin and Its Receptor System in Colorectal Cancer. Biomedicines 2021, 9, 1743.
- de Bruïne, A.P.; Wiggers, T.; Beek, C.; Volovics, A.; von Meyenfeldt, M.; Arends, J.W.; Bosman, F.T. Endocrine cells in colorectal adenocarcinomas: Incidence, hormone profile and prognostic relevance. Int. J. Cancer 1993, 54, 765–771.
- de Bruïne, A.P.; Dinjens, W.N.; Pijls, M.M.; vd Linden, E.P.; Rousch, M.J.; Moerkerk, P.T.; de Goeij, A.F.; Bosman, F.T. NCI-H716 cells as a model for endocrine differentiation in colorectal cancer. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1992, 62, 311–320.
- Godlewski, J.; Kaleczyc, J. Somatostatin, substance P and calcitonin gene-related peptide-positive intramural nerve structures of the human large intestine affected by carcinoma. Folia Histochem. Cytobiol. 2010, 48, 475–483.
- El-Salhy, M.; Mahdavi, J.; Norrgård, O. Colonic endocrine cells in patients with carcinoma of the colon. Eur. J. Gastroenterol. Hepatol. 1998, 10, 517–522.
- Sereti, E.; Gavriil, A.; Agnantis, N.; Golematis, V.C.; Voloudakis-Baltatzis, I.E. Immunoelectron study of somatostatin, gastrin and glucagon in human colorectal adenocarcinomas and liver metastases. Anticancer Res. 2002, 22, 2117–2123.
- Seretis, E.C.; Agnantis, N.J.; Golematis, V.C.; Voloudakis-Balatzis, I.E. Electron immunocytochemical demonstration of serotonin, vasoactive intestinal polypeptide, bombesin, somatostatin and glucagon in mirror biopsies from primary colorectal adenocarcinoma. J. Exp. Clin. Cancer Res. 2004, 23, 477–484.
- Leiszter, K.; Sipos, F.; Galamb, O.; Krenács, T.; Veres, G.; Wichmann, B.; Fűri, I.; Kalmár, A.; Patai, Á.V.; Tóth, K.; et al. Promoter hypermethylation-related reduced somatostatin production promotes uncontrolled cell proliferation in colorectal cancer. PLoS ONE 2015, 10, e0118332.
- Seretis, E.; Konstantinidou, A.; Arnogiannakis, N.; Xinopoulos, D.; Voloudakis-Baltatzis, I.E. Mucinous colorectal adenocarcinoma with signet-ring cells: Immunohistochemical and ultrastructural study. Ultrastruct. Pathol. 2010, 34, 337–343.
- Sitohy, B.; El-Salhy, M. Colonic endocrine cells in rats with chemically induced colon carcinoma. Histol. Histopathol. 2001, 16, 833–838.
- Yao, G.Y.; Zhou, J.L.; Lai, M.D.; Chen, X.Q.; Chen, P.H. Neuroendocrine markers in adenocarcinomas: An investigation of 356 cases. World J. Gastroenterol. 2003, 9, 858–861.
- Hameed, Y.; Ahmad, M.; Ejaz, S.; Liang, S. Identification of Key Biomarkers for the Future Applications in Diagnostics and Targeted Therapy of Colorectal Cancer. Curr. Mol. Med. 2022, 22, 31–37.
- Iftikhar, S.Y.; Watson, S.A.; Morris, D.L. The effect of long acting somatostatin analogue SMS 201.995 therapy on tumour kinetic measurements and serum tumour marker concentrations in primary rectal cancer. Br. J. Cancer 1991, 63, 971–974.
- Stewart, G.J.; Connor, J.L.; Lawson, J.A.; Preketes, A.; King, J.; Morris, D.L. Octreotide reduces the kinetic index, proliferating cell nuclear antigen-maximum proliferative index, in patients with colorectal cancer. Cancer 1995, 76, 572–578.
- Pollak, M.N.; Polychronakos, C.; Guyda, H. Somatostatin analogue SMS 201-995 reduces serum IGF-I levels in patients with neoplasms potentially dependent on IGF-I. Anticancer Res. 1989, 9, 889–891.
- Klijn, J.G.; Hoff, A.M.; Planting, A.S.; Verweij, J.; Kok, T.; Lamberts, S.W.; Portengen, H.; Foekens, J.A. Treatment of patients with metastatic pancreatic and gastrointestinal tumours with the somatostatin analogue Sandostatin: A phase II study including endocrine effects. Br. J. Cancer 1990, 62, 627–630.
- Di Leo, A.; Bajetta, E.; Ferrari, L.; Biganzoli, L.; Mariani, L.; Carnaghi, C.; Camerini, E.; Buzzoni, R.; Ruiz, J.M. A dose-finding study of lanreotide (a somatostatin analog) in patients with colorectal carcinoma. Cancer 1996, 78, 35–42.
- Cascinu, S.; Del Ferro, E.; Grianti, C.; Ligi, M.; Ghiselli, R.; Foglietti, G.; Saba, V.; Lungarotti, F.; Catalano, G. Inhibition of tumor cell kinetics and serum insulin growth factor I levels by octreotide in colorectal cancer patients. Gastroenterology 1997, 113, 767–772.
- Wu, P.; Mao, J.D.; Yan, J.Y.; Rui, J.; Zhao, Y.C.; Li, X.H.; Xu, G.Q. Correlation between the expressions of gastrin, somatostatin and cyclin and cyclin-depend kinase in colorectal cancer. World J. Gastroenterol. 2005, 11, 7211–7217.
- Fogh, J.; Trempe, G. New human tumor cell lines. In Human Tumor Cells In Vitro; Fogh, J., Ed.; Springer: Boston, MA, USA, 1975; pp. 115–159.
- Staykova, S.T.; Wesselinova, D.W.; Vezenkov, L.T.; Naydenova, E.D. Synthesis and in vitro antitumor activity of new octapeptide analogs of somatostatin containing unnatural amino acids. Amino Acids. 2015, 47, 1007–1013.
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71.
- Olejniczak, A.; Szaryńska, M.; Kmieć, Z. In vitro characterization of spheres derived from colorectal cancer cell lines. Int. J. Oncol. 2018, 52, 599–612.
- Brattain, M.G.; Fine, W.D.; Khaled, F.M.; Thompson, J.; Brattain, D.E. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981, 41, 1751–1756.
- Smith, J.P.; Solomon, T.E. Effects of gastrin, proglumide, and somatostatin on growth of human colon cancer. Gastroenterology 1988, 95, 1541–1548.
- Björk, J.; Nilsson, J.; Hultcrantz, R.; Johansson, C. Growth-regulatory effects of sensory neuropeptides, epidermal growth factor, insulin, and somatostatin on the non-transformed intestinal epithelial cell line IEC-6 and the colon cancer cell line HT 29. Scand. J. Gastroenterol. 1993, 28, 879–884.
- Radulovic, S.; Schally, A.V.; Reile, H.; Halmos, G.; Szepeshazi, K.; Groot, K.; Milovanovic, S.; Miller, G.; Yano, T. Inhibitory effects of antagonists of bombesin/gastrin releasing peptide (GRP) and somatostatin analog (RC-160) on growth of HT-29 human colon cancers in nude mice. Acta Oncol. 1994, 33, 693–701.
- Tóvári, J.; Szende, B.; Bocsi, J.; Falaschi, A.; Simoncsits, A.; Pongor, S.; Erchegyi, J.; Steták, A.; Kéri, G. A somatostatin analogue induces translocation of Ku 86 autoantigen from the cytosol to the nucleus in colon tumour cells. Cell Signal. 1998, 10, 277–282.
- Tejeda, M.; Gaál, D.; Hullán, L.; Hegymegi-Barakonyi, B.; Kéri, G. Evaluation of the antitumor efficacy of the somatostatin structural derivative TT-232 on different tumor models. Anticancer Res. 2006, 26, 3477–3483.
- Staykova, S.; Naydenova, E.; Wesselinova, D.; Kalistratova, A.; Vezenkov, L. Synthesis and in vitro study of the anticancer activity of new analogs of octreotide. Protein Pept. Lett. 2012, 19, 1257–1262.
- Ayiomamitis, G.D.; Notas, G.; Zaravinos, A.; Drygiannakis, I.; Georgiadou, M.; Sfakianaki, O.; Mastrodimou, N.; Thermos, K.; Kouroumalis, E. Effects of octreotide and insulin on colon cancer cellular proliferation and correlation with hTERT activity. Oncoscience 2014, 1, 457–467.
- Naydenova, E.; Wesselinova, D.; Staykova, S.; Danalev, D.; Dzimbova, T. Synthesis, in vitro biological activity and docking of new analogs of BIM-23052 containing unnatural amino acids. Amino Acids. 2019, 51, 1247–1257.
- Kéri, G.; Erchegyi, J.; Horváth, A.; Mezõ, I.; Idei, M.; Vántus, T.; Balogh, A.; Vadász, Z.; Bökönyi, G.; Seprõdi, J.; et al. A tumor-selective somatostatin analog (TT-232) with strong in vitro and in vivo antitumor activity. Proc. Natl. Acad. Sci. USA 1996, 93, 12513–12518.
- Dy, D.Y.; Whitehead, R.H.; Morris, D.L. SMS 201.995 inhibits in vitro and in vivo growth of human colon cancer. Cancer Res. 1992, 52, 917–923.
- di Paolo, A.; Bocci, G.; Innocenti, F.; Agen, C.; Nardini, D.; Danesi, R.; del Tacca, M. Inhibitory effect of the somatostatin analogue SMS 201-995 and cytokines on the proliferation of human colon adenocarcinoma cell lines. Pharmacol. Res. 1995, 32, 135–139.
- Vántus, T.; Csermely, P.; Teplán, I.; Kéri, G. The tumor-selective somatostatin analog, TT2-32 induces a biphasic activation of phosphotyrosine phosphatase activity in human colon tumor cell line, SW620. Tumour Biol. 1995, 16, 261–267.
- Hohla, F.; Buchholz, S.; Schally, A.V.; Krishan, A.; Rick, F.G.; Szalontay, L.; Papadia, A.; Halmos, G.; Koster, F.; Aigner, E.; et al. Targeted cytotoxic somatostatin analog AN-162 inhibits growth of human colon carcinomas and increases sensitivity of doxorubicin resistant murine leukemia cells. Cancer Lett. 2010, 294, 35–42.
- Qin, Y.; Schally, A.V.; Willems, G. Somatostatin analogue RC-160 inhibits the growth of transplanted colon cancer in rats. Int. J. Cancer. 1991, 47, 765–770.
- Romani, R.; Morris, D.L. SMS 201.995 (Sandostatin) enhances in-vitro effects of 5-fluorouracil in colorectal cancer. Eur. J. Surg. Oncol. 1995, 21, 27–32.
- Ogasawara, M.; Murata, J.; Ayukawa, K.; Saiki, I. Differential effect of intestinal neuropeptides on invasion and migration of colon carcinoma cells in vitro. Cancer Lett. 1997, 119, 125–130.
- Chen, J.S.; Liang, Q.M.; Li, H.S.; Yang, J.; Wang, S.; Long, J.W. Octreotide inhibits growth of colonic cancer SW480 cells by modulating the Wnt/P-catenin pathway. Pharmazie 2009, 64, 126–131.
- Szepeshazi, K.; Schally, A.V.; Halmos, G.; Armatis, P.; Hebert, F.; Sun, B.; Feil, A.; Kiaris, H.; Nagy, A. Targeted cytotoxic somatostatin analogue AN-238 inhibits somatostatin receptor-positive experimental colon cancers independently of their p53 status. Cancer Res. 2002, 62, 781–788.
- Tjomsland, V.; El-Salhy, M. Anti-tumour effects of triple therapy with octreotide, galanin and serotonin in comparison with those of 5-fluorouracil/leukovorin on human colon cancer. Int. J. Oncol. 2005, 27, 427–432.
- El-Salhy, M. Effects of triple therapy with octreotide, galanin and serotonin on a human colon cancer cell line. Oncol. Rep. 2005, 13, 45–49.
- Szegedi, Z.; Takács, J.; Szende, B.; Vadász, Z.; Horváth, A.; Gulyás, E.; Tóth, G.; Peták, I.; Bocsi, J.; Kéri, G. A specifically radiolabeled somatostatin analog with strong antitumor activity induces apoptosis and accumulates in the cytosol and the nucleus of HT29 human colon carcinoma cells. Endocrine 1999, 10, 25–34.
- Colucci, R.; Blandizzi, C.; Ghisu, N.; Florio, T.; Del Tacca, M. Somatostatin inhibits colon cancer cell growth through cyclooxygenase-2 downregulation. Br. J. Pharmacol. 2008, 155, 198–209.
- Massari, D.; Trobonjac, Z.; Rukavina, D.; Radosević-Stasić, B. SMS 201-995 enhances S-phase block induced by 5-fluorouracil in a human colorectal cancer cell line. Anticancer Drugs. 2005, 16, 989–996.
- Abdellatif, A.A.H.; Ibrahim, M.A.; Amin, M.A.; Maswadeh, H.; Alwehaibi, M.N.; Al-Harbi, S.N.; Alharbi, Z.A.; Mohammed, H.A.; Mehany, A.B.M.; Saleem, I. Cetuximab Conjugated with Octreotide and Entrapped Calcium Alginate-beads for Targeting Somatostatin Receptors. Sci. Rep. 2020, 10, 4736.
- Fan, J.; Deng, L.; Peng, Y.; Ding, Y. Combined anti-tumor efficacy of somatostatin fusion protein and vaccinia virus on tumor cells with high expression of somatostatin receptors. Sci. Rep. 2022, 12, 16885.
- den Hertog, J.; Ostman, A.; Böhmer, F.D. Protein tyrosine phosphatases: Regulatory mechanisms. FEBS J. 2008, 275, 831–847.
- Kéri, G.; Mezö, I.; Horváth, A.; Vadász, Z.; Balogh, A.; Idei, M.; Vántus, T.; Teplán, I.; Mák, M.; Horváth, J.; et al. Novel somatostatin analogs with tyrosine kinase inhibitory and antitumor activity. Biochem. Biophys. Res. Commun. 1993, 191, 681–687.
- Kéri, G.; Mezô, I.; Vadász, Z.; Horváth, A.; Idei, M.; Vántus, T.; Balogh, A.; Bökönyi, G.; Bajor, T.; Teplán, I.; et al. Structure-activity relationship studies of novel somatostatin analogs with antitumor activity. Pept. Res. 1993, 6, 281–288.
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184.
- Neel, B.G. Structure and function of SH2-domain containing tyrosine phosphatases. Semin. Cell Biol. 1993, 4, 419–432.
- Bertorelle, R.; Rampazzo, E.; Pucciarelli, S.; Nitti, D.; De Rossi, A. Telomeres, telomerase and colorectal cancer. World J. Gastroenterol. 2014, 20, 1940–1950.
- Le Romancer, M.; Reyl-Desmars, F.; Cherifi, Y.; Pigeon, C.; Bottari, S.; Meyer, O.; Lewin, M.J. The 86-kDa subunit of autoantigen Ku is a somatostatin receptor regulating protein phosphatase-2A activity. J. Biol. Chem. 1994, 269, 17464–17468.
- Pucci, S.; Bonanno, E.; Pichiorri, F.; Mazzarelli, P.; Spagnoli, L.G. The expression and the nuclear activity of the caretaker gene ku86 are modulated by somatostatin. Eur. J. Histochem. 2004, 48, 103–110.
- Pucci, S.; Mazzarelli, P.; Sesti, F.; Boothman, D.A.; Spagnoli, L.G. Interleukin-6 affects cell death escaping mechanisms acting on Bax-Ku70-Clusterin interactions in human colon cancer progression. Cell Cycle. 2009, 8, 473–481.
- Saydam, O.; Saydam, N. Deficiency of Ku Induces Host Cell Exploitation in Human Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 651818.
- Dy, D.Y.; Morris, D.L. Somatostatin inhibits both in-vitro and in-vivo carcinoembryonic antigen secretion by human colon cancer. Eur. J. Surg. Oncol. 1993, 19, 168–172.
- Mao, J.D.; Wu, P.; Xia, X.H.; Hu, J.Q.; Huang, W.B.; Xu, G.Q. Correlation between expression of gastrin, somatostatin and cell apoptosis regulation gene bcl-2/bax in large intestine carcinoma. World J. Gastroenterol. 2005, 11, 721–725.
- Mao, J.D.; Wu, P.; Yang, Y.L.; Wu, J.; Huang, H. Relationship between expression of gastrin, somatostatin, Fas/FasL and caspases in large intestinal carcinoma. World J. Gastroenterol. 2008, 14, 2802–2809.
- Mełen-Mucha, G.; Winczyk, K.; Pawlikowski, M. Somatostatin analogue octreotide and melatonin inhibit bromodeoxyuridine incorporation into cell nuclei and enhance apoptosis in the transplantable murine colon 38 cancer. Anticancer Res. 1998, 18, 3615–3619.
- Wang, S.; Bao, Z.; Liang, Q.M.; Long, J.W.; Xiao, Z.S.; Jiang, Z.J.; Liu, B.; Yang, J.; Long, Z.X. Octreotide stimulates somatostatin receptor-induced apoptosis of SW480 colon cancer cells by activation of glycogen synthase kinase-3β, A Wnt/β-catenin pathway modulator. Hepatogastroenterology 2013, 60, 1639–1646.
- El-Salhy, M.; Sitohy, B.; Norrgård, O. Triple therapy with octreotide, galanin, and serotonin reduces the size and blood vessel density and increases apoptosis of a rat colon carcinoma. Regul. Pept. 2003, 111, 145–152.
- El-Salhy, M. Comparison between triple therapy with octreotide, galanin and serotonin, 5-fluorouracil/leucovorin, and sequential treatment with both, on human colon cancer. Oncol. Rep. 2004, 11, 1161–1168.
- El-Salhy, M.; Dennerqvist, V. Effects of triple therapy with octreotide, galanin and serotonin on liver metastasis of human colon cancer in xenografts. Oncol. Rep. 2004, 11, 1177–1182.
- El-Salhy, M. Triple treatment with octreotide, galanin and serotonin is a promising therapy for colorectal cancer. Curr. Pharm. Des. 2005, 11, 2107–2117.
- El-Salhy, M.; Hilding, L.; Royson, H.; Tjomsland, V. Comparison between triple therapy with octreotide, galanin and serotonin vs. irinotecan or oxaliplatin in combination with 5-fluorouracil/leukovorin in human colon cancer. Int. J. Oncol. 2005, 27, 687–691.
- Jaggi, M.; Prasad, S.; Singh, A.T.; Praveen, R.; Dutt, S.; Mathur, A.; Sharma, R.; Gupta, N.; Ahuja, R.; Mukherjee, R.; et al. Anticancer activity of a peptide combination in gastrointestinal cancers targeting multiple neuropeptide receptors. Investig. New Drugs. 2008, 26, 489–504.
- Singh, A.T.; Jaggi, M.; Prasad, S.; Dutt, S.; Singh, G.; Datta, K.; Rajendran, P.; Sanna, V.K.; Mukherjee, R.; Burman, A.C. Modulation of key signal transduction molecules by a novel peptide combination effective for the treatment of gastrointestinal carcinomas. Investig. New Drugs. 2008, 26, 505–516.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
627
Revisions:
2 times
(View History)
Update Date:
13 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No