Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kuldeep Tripathi | -- | 3148 | 2024-03-07 20:22:55 | | | |
| 2 | Wendy Huang | Meta information modification | 3148 | 2024-03-08 04:55:11 | | | | |
| 3 | Franklin Nnachetam Aneke | Meta information modification | 3148 | 2024-03-08 09:16:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tripathi, K.; Ben-Shachar, D.; Franklin, A.N. Mitochondria in the Central Nervous System Disorder. Encyclopedia. Available online: https://encyclopedia.pub/entry/55989 (accessed on 26 July 2026).
Tripathi K, Ben-Shachar D, Franklin AN. Mitochondria in the Central Nervous System Disorder. Encyclopedia. Available at: https://encyclopedia.pub/entry/55989. Accessed July 26, 2026.
Tripathi, Kuldeep, Dorit Ben-Shachar, Aneke Nnachetam Franklin. "Mitochondria in the Central Nervous System Disorder" Encyclopedia, https://encyclopedia.pub/entry/55989 (accessed July 26, 2026).
Tripathi, K., Ben-Shachar, D., & Franklin, A.N. (2024, March 07). Mitochondria in the Central Nervous System Disorder. In Encyclopedia. https://encyclopedia.pub/entry/55989
Tripathi, Kuldeep, et al. "Mitochondria in the Central Nervous System Disorder." Encyclopedia. Web. 07 March, 2024.
Copy Citation
Mitochondria, the energy suppliers of the cells, play a central role in a variety of cellular processes essential for survival or leading to cell death. Consequently, mitochondrial dysfunction is implicated in numerous general and central nervous system (CNS) disorders. The clinical manifestations of mitochondrial dysfunction include metabolic disorders, dysfunction of the immune system, tumorigenesis, and neuronal and behavioral abnormalities.
mitochondria
CNS
mitochondrial transplantation
CNS disorders
dysfunction
1. Introduction
The traditional view of the mitochondria, the double-membrane organelle, was based on their central role in cellular respiration, a process that converts nutrients into adenosine triphosphate (ATP), the primary energy currency of the cell. At present, mitochondria are conceptualized as a cellular hub that maintains cellular homeostasis, regulates apoptosis, calcium signaling, and cellular oxidative stress states [1][2][3]. In addition, the mitochondrial tricarboxylic acid (TCA) cycle provides metabolites that serve as building blocks for a variety of macromolecules including proteins, lipids, carbohydrates, and nucleotides. Mitochondria also control the innate immune response and are essential mediators of epigenetic processes [4] (Figure 1). Serving as a hub, the mitochondria respond to the cellular state and its energy demands, which are transmitted by cytosolic signaling molecules such as Ca2+ and protein kinases and by neurotransmitters [5][6][7][8][9][10]. Maintaining overall cellular and mitochondrial homeostasis is strongly dependent on the coordination between mitochondrial and nuclear DNA [11]. The nuclear genes coordinate the synthesis of proteins essential for mitochondrial structure and function and both nuclear and mitochondrial DNA-encoded factors are involved in the regulation of mitochondrial DNA replication and repair [12].
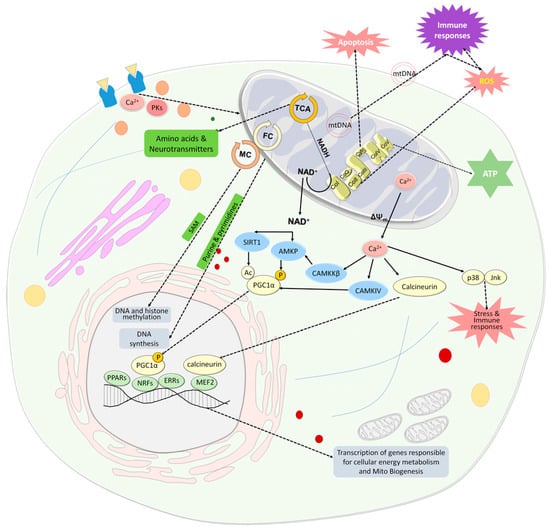
Figure 1. Mitochondria are an important cellular hub. Mitochondria produce energy in the form of ATP through the OxPhos system, which is also a major source of ROS and is involved in apoptosis. The TCA cycle not only provides reduced substrates in the form of NADH and NADPH for the respiratory chain, but also provides amino acids, that are converted to neurotransmitters and serve as building blocks for proteins. The folate cycle, which is located in the mitochondria and the cytosol, provides purines and pyrimidines for DNA synthesis and, together with the methionine cycle, provides methyl groups for DNA and histone methylation. In addition, mitochondria communicate with the nucleus in homeostasis and stress. Changes in the NAD+/NADH ratio and extracellular Ca2+ concentrations are two major inducers of signaling pathways that activate PGC-1α. An increase in NAD+ levels is associated with reduced ATP levels and increased extracellular Ca2+ concentration is linked to dissipation of ΔΨm and enhanced leak of mitochondrial Ca2+. NAD+ activates SIRT1 which in turn activates PGC-1α by its deacetylation. NAD+ also activates AMKP which in turn activates PGC-1α by its phosphorylation. AMPK can also be activated by CaMKKβ, which is activated by Ca2+. PGC-1α is an upstream inducer of mitochondrial genes. It positively affects the activity of some hormone nuclear receptors, PPARs and ERRs, which regulate multiple pathways involved in cellular energy metabolism within and outside mitochondria, and nuclear transcription factors, NRFs, which leads to the transcription of nuclear coded respiratory chain components and Tfam. PGC1α can be also activated by CaMKIV which in turn is activated by Ca2+. Ca2+ also activates the phosphatase calcineurin, which activates the nuclear MEF2, which in turn regulates many muscle-specific genes and lipogenic and glycogenic enzyme genes. Ca2+ released from the mitochondria induces a stress state via the activation of stress-related MAPKs, p38 and Jnk, which leads to the activation of the immune system and oxidative stress state as do cell-free mtDNAs. Mitochondria are also involved in hemoglobin and steroid production and respond to the cellular state and its energy demands, transmitted by cytosolic signaling molecules such as Ca2+ and protein kinases and by neurotransmitters. Abbreviations: Ac—acetyl group; AMPK—AMP-activated protein kinase; CaMKIV—Ca2+/calmodulin-dependent protein kinase type IV; CaMKKβ—Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ); Co—complex; ERRs—estrogen-related receptors; FC—folate cycle; Jnk—c-Jun NH2-terminal kinase; MC—methionine cycle; MEF2—myocyte-specific enhancer factor 2; mtDNA- mitochondrial DNA; NAD—nicotinamide adenine dinucleotide; NFRs—nuclear respiratory factors; OxPhos-oxidative phosphorylation; P—phosphate group; p38—p38 mitogen-activated protein kinase; PGC-1α—PPARγ co-activator 1α; PK—protein kinases; PPARs—peroxisome proliferator-activated receptors; ROS—reactive oxygen species; SAM—S-adenosylmethionine; TCA—tricarboxylic acid; Tfam—mitochondria transcription factor A; ΔΨm—mitochondrial membrane potential.
2. Mitochondria and CNS Diseases
Forming such an important cellular hub and being essential players in cellular survival and death, it is comprehensible that mitochondria are strongly involved in pathological processes leading to numerous general and CNS diseases. Mitochondrial malfunction can be attributed to multiple mitochondrial factors including their enzymes’ function and their mtDNA. Mitochondria in cells are not a homogenous population. A recent study showed, for the first time, two distinct populations of mitochondria in rat liver, cytoplasmic mitochondria and lipid droplet-associated mitochondria. The lipid droplet mitochondria were more efficient in fatty acid oxidation and exhibited specific molecular markers such as carnitine palmitoyl transferase (CPT1), phosphorylated acetyl- CoA carboxylase (ACC), and MFN2, while cytoplasmic mitochondria had higher respiration capacity. Interestingly, mitochondria associated with lipid droplets in a non-alcoholic fatty liver disease (NAFLD) rat model showed compromised fatty acid oxidation, highlighting the role of droplet-associated mitochondria in NAFLD and the significance of functional separation of mitochondria to treat NAFLD [13] and possibly other disorders.
The essential role of mitochondria in neurons has been described above. Any distortion in their activity can lead to changes in synaptic plasticity and neuronal network connectivity, eventually resulting in behavioral changes and neurodegeneration. Astrocytes have a crucial role in supporting neurons, and changes in their mitochondrial function are linked to the maintenance of brain health. Astrocytes typically have low activity of mitochondrial OxPhos as compared to neurons. Nevertheless, it was demonstrated that their OxPhos is essential for degrading fatty acids and maintaining lipid balance in the brain. Astrocytic OxPhos malfunctions can cause accumulation of lipid droplets and elevated acetyl-CoA levels, inducing astrocyte reactivity. The astrocytes’ lipid-centric mechanism was shown to stimulate neuronal fatty acid oxidation and oxidative stress, to activate microglia, and hinder the synthesis of essential lipids for myelin replenishment. As a consequence synaptic loss, demyelination, and neuroinflammation were observed, leading to cognitive decline and neurodegeneration relevant to AD and maybe also to multiple sclerosis (MS) [14]. Ample evidence from genetic, transcriptomic, and proteomic studies supports an association between mitochondrial enzyme deficiencies, specifically those of the OxPhos, and the pathophysiology of CNS disorders, including mental illness, such as schizophrenia, major depression, and bipolar disorders as well as neurodegenerative disorders such as Parkinson’s disease (PD) and AD. For example, in schizophrenia, major depression, and bipolar disorder, a deficiency in the first complex of the OxPhos system, NADH/ubiquinone oxidoreductase (complex I) is repeatedly reported, while in PD and AD, as well as in aging, a deficit is observed in both complex I and the fourth complex, cytochrome-c-oxidase [15][16][17][18][19][20][21][22][23][24].
Mitochondrial TCA cycle intermediates have an essential role in the synthesis of various biomolecules and signaling molecules, which are involved in chromatin modifications, DNA methylation, immunity, and hypoxic response (for a review see [25]). Hence, TCA cycle enzymes have been associated with various CNS dysfunctions. For example, in schizophrenia, enzymes of the first half of the TCA cycle (aconitase, alpha-ketoglutarate dehydrogenase complex, and succinyl coenzyme A synthetase) showed lower activity, while those in the second half (succinate dehydrogenase and malate dehydrogenase) showed higher activities, probably compensating for the lower activity levels of the first half of the cycle [26]. In AD, deficits in brain TCA cycle enzymes, specifically pyruvate dehydrogenase subunit beta (PDHB), succinate-CoA ligase [ADP-Forming] Subunit Beta (SUCLA2), and malate dehydrogenase 1 (MDH1) were observed [27]. Finally, patients with inherited deficiencies in TCA enzymes such as fumarate hydratase (FH) and α-ketoglutarate dehydrogenase (α-KGDH) present, among other symptoms, progressive and severe encephalopathy, psychiatric and pyramidal symptoms, and developmental delay [28][29].
mtDNA also contributes to CNS diseases, mainly due to the strong inflammation associated with increased circulating cell-free (cf)-mtDNA levels (Figure 1). Similar to cf-nuclear DNA [30], the role of cf-mtDNA is increasingly recognized in immune-mediated systemic and central inflammatory diseases [31][32]. The measurement of cf-mtDNA concentration emerges as a potential biomarker for acute inflammatory stress. Damaged mtDNA released into the cytosol activates inflammatory pathways, including the inflammasome and NF-κB, contributing to inflammation at various levels [33][34]. Notably, elevated levels of mtDNA are observed in plasma in CNS disorders with strong inflammatory responses, such as relapsing–remitting multiple sclerosis [35][36]. An increase in cf-mtDNA may reflect early inflammatory activity, leading to mitochondrial damage, neural loss, and brain atrophy and its change has been suggested as a predictor for treatment outcome [37][38]. Interestingly, a reduction in melatonin, an antioxidant produced by neurons and shown to be involved in mitochondrial homeostasis, can lead to disruption of mitochondria, the release of mtDNA, and activation of a cytosolic inflammatory response in neurons, by triggering the cGAS/STING/IRF3 pathway [39]. In PD, it has been suggested that mitochondrial dysfunction can lead to the release of mitochondrial damage-associated molecular patterns (mtDAMPs), triggering neuroinflammation through microglial immune receptors. Conversely, inflammatory molecules released by the glial cells can further damage mitochondrial function, triggering a vicious circle that can lead to neurodegeneration [40][41]. In mental disorders such as major depression and schizophrenia, higher levels of cf-mtDNA were associated with symptoms of depression and schizophrenia. Concomitantly, treatment with certain mood stabilizers (lamotrigine, valproic acid, or lithium) was associated with lower cf-mtDNA [42][43][44][45]. In addition to alterations in cf-mtDNA, changes in the mtDNA genome were also shown to be associated with CNS disorders. For example, SNPs in two control regions in the mtDNA (T16519C and T195C) as well as in mtDNA-encoded cytochrome B (CYTB), which protein is a component of the ubiquinol–cytochrome c reductase complex (complex III), were associated with schizophrenia and bipolar disorders. A12308G was associated with psychosis in both disorders [46]. Several point mtDNA mutation deletions and methylation, as well as changes in mtDNA copy numbers, have been associated with neurodegenerative disorders including PD and AD, amyotrophic lateral sclerosis, and Huntington’s disease (for a review see [47]). In monozygotic twins discordant for schizophrenia, a discordance in mtDNA copy number estimates was observed suggesting post-zygotic mtDNA changes that may contribute to the discordance in the disease between monozygotic twins [48]. mtDNA copy number in peripheral blood cells has been suggested as a potential biomarker as it was reduced in PD, increased in attention-deficit hyperactivity disorder (ADHD), and associated with psychosis severity and antipsychotic treatment in schizophrenia [49][50][51]. Taken together, all these findings corroborate that mitochondrial multifaceted dysfunction contributes to the pathogenesis of CNS disorders.
3. Mitochondrial Transplantation in CNS Disorder Models
During the past decade, studies examining the therapeutic potential of transplantation of isolated extracellular mitochondria in a variety of general and CNS disease models have thrived (for reviews see [52][53][54][55] and Table 1). The beneficial effects of exogenous mitochondrial transplantation have been demonstrated in diverse pathological models including cardiac, lung, liver, and CNS pathologies [56][57][58][59][60][61][62][63]. In humans, a few clinical trials showed beneficial effects of autologous mitochondrial transplantation, but the follow-up period was relatively short. In infants who required ECMO support for ischemia–reperfusion-associated myocardial dysfunction, autologous mitochondrial transplantation led to an improvement in ventricular function and release from ECMO support. However, there is no information on how stable the improvement was [64]. Another human study was performed on six children with single large-scale mitochondrial DNA (mtDNA) de novo deletion syndromes (SLSMDs), a rare and severe multisystemic disease. Mitochondrial augmentation therapy, in which the maternal mitochondria, mostly similar to those of the recipient, were transplanted into the patients’ enriched hematopoietic cells following leukapheresis, was employed. Following the transplantation procedure, partial and limited improvement in mitochondrial number and clinical symptoms was observed during 6–12 months of follow-up [65]. In women with recurrent pregnancy failures, transplantation of autologous mitochondria to mature human oocytes with sperm at the time of intracytoplasmic sperm injection resulted in a significant improvement in the ratio of good-quality embryos and healthy normal babies [66]. All of these studies used autologous or allogeneic maternal-derived mitochondria for transplantation rather than allogeneic mitochondria to reduce immune response and avoid heteroplasmy.
Several studies have transplanted exogenous mitochondria in various CNS experimental models. In 6-OHDA Parkinson’s disease rat model, transplantation of exogenous allogeneic and xenogeneic mitochondria coupled with Pep-1 into the medial forebrain bundle (MFB) reduced loss of dopaminergic neurons in the substantia nigra three months later, suggesting that the effect of the transplanted mitochondria spread beyond the injection site, probably due to the characteristics of the MFB, being a neural pathway containing fibers. The restoration of the dopaminergic neurons was associated with enhanced mitochondrial functions, reduced neuroinflammation, and enhanced locomotive activity [53]. In this study, no significant difference was observed between Pep-1-coupled xenogeneic and allogeneic mitochondria-induced effects. The same group showed similar beneficial bioenergetical and behavioral effects at three to four weeks following repeated chronic (once a week for three months) intranasal injection of Pep-1-labeled mitochondria [63]. Another study used the MPTP null mouse model of PD and intravenously injected them with exogenous mitochondria. The GFP-labeled transplanted mitochondria were distributed in various organs including the liver, kidney, muscle, and brain. MPTP mice systemically injected with mitochondria showed improved locomotion, reduced ROS generation, and restored ATP levels and complex I activity. Systemically injected mitochondria in healthy mice did not affect ATP levels nor spontaneous locomotion but significantly increased latency to immobility in the forced swimming test [67]. Both studies showed improvements in cell survival and mitochondrial functions in cell cultures treated with the relevant toxins. Injection of exogenous mitochondria into the tail vein was also performed in a mouse model of AD, produced by intracerebroventricular injection with amyloid-beta 1–42 peptide. One to two weeks after the intravenous injections of fresh human-isolated mitochondria, AD mice exhibited significantly improved cognitive performance. Furthermore, there was a notable reduction in neuronal loss and gliosis in the hippocampus and increased activities of citrate synthase and cytochrome c oxidase in treated AD mice compared to non-treated AD mice [60]. Acute effects of intravenous repeated transplantation of mitochondria isolated from young mice into aged mice included improved mitochondrial bioenergetics and reduced redox state, associated with ameliorated learning and motor functions in the aged mice [68]. The protective effect of exogenous mitochondrial transplantation was also studied in experimental spinal cord injury (SCI). One study reported that up to 7 days post-transplantation, mitochondria were observed in multiple resident cell types but not in neurons. The other study detected the transplanted mitochondria up to 28 days post-transplantation in the vicinity of the lesion. One day after transplantation, a partial restoration of mitochondrial respiration as well as amelioration of mitochondrial fragmentation and cellular apoptosis were observed. Partial functional protection, assessed by tissue paring and recovery of sensory and motor function, was observed four weeks after transplantation; however, it faded after 6 weeks of follow-up [69][70]. In traumatic brain injury, which caused mitochondrial impairments, anxiety and cognitive deficits, transplantation of allogeneic liver-isolated mitochondria restored astrocytic brain-derived neurotrophic factor (BDNF) levels and the behavioral deficits [71].
The past decade has witnessed an abundance of studies focusing on mitochondrial abnormalities in several mental disorders including major depression and schizophrenia. A wide array of methodologies ranging from imaging through genetic, biochemical, and molecular to histological and structural techniques were used to reveal multifaceted mitochondrial dysfunction in mental disorders, specifically in schizophrenia [72][73][74]. Hence, mitochondrial transplantation effects were also assessed in experimental models of mental disorders. In a lipopolysaccharide-induced mouse model of inflammation-induced depression, intravenous injection of exogenous mitochondria acutely reduced depressive-like behaviors assessed by forced swim, tail suspension, and sucrose preference tests. This was associated with an acute reduction in astrocyte and microglia activation and cytokines levels, higher levels of BDNF transcripts and neurogenesis, and restored mitochondrial dysfunction measured by ATP and ROS production in mouse hippocampi [75]. We have studied the effect of exogenous allogeneic mitochondrial transplantation in schizophrenia patient-derived iPSCs (SZ-iPSCs) and in the maternal immune activation Poly I:C rat model of schizophrenia. Transplantation of mitochondria into SZ-iPSCs restored mitochondrial deficits including mitochondria respiration, ΔΨm, mitochondrial network dynamics, and transcript levels of specific subunits of complex I and of OPA1. Concomitantly, an enhanced efficiency of SZ-iPSCs differentiating into functional dopaminergic and glutamatergic neurons was observed. In the rat model of SZ, intra-medial prefrontal cortex (mPFC) single injection of mitochondria, in adolescent rats (34 days old), restored mitochondrial impairments and neuronal outgrowth and activity. assessed by monoamines’ transmission. in adulthood (100–120 days old). Proteomics analysis of the mPFC showed an association between the beneficial neuronal and mitochondrial effects and improved metabolic and neuronal development and plasticity pathways. Finally, mitochondrial transplantation in Poly I:C rats restored schizophrenia-related behavioral deficits such as attentional deficit and spontaneous locomotor activity in a novel environment. The behavioral changes showed a significant correlation with changes in monoamine and neuronal structural alterations. Unlike the intra-MFB injection in the PD model, the data suggest a localized effect of the transplanted mitochondria. Unexpectedly, a similar injection protocol to healthy rats induced detrimental effects in all parameters mentioned above, including behavioral, bioenergetical, and neuronal-related features. These data emphasize the importance of the bioenergetical and physiological states of the recipient in the outcome of mitochondrial transplantation. Furthermore, the opposite effects induced by mitochondrial transplantation in the schizophrenia model and healthy rats advocate for a causal link between the mitochondria and behavior, neuronal activity and plasticity [61][62].
Table 1. Research reports on mitochondrial transplantation in CNS disease models.
| Disease | Species/Applied Model | Source of Mitochondria | Route of Transplantation | Ref. |
|---|---|---|---|---|
| Parkinson’s disease | Rat | Allogeneic/xenogeneic Pep-1-labeled mitochondria | Injection into the medial forebrain bundle | [76] |
| Parkinson’s disease | Mouse | Human hepatoma cells | Intravenous injection | [67] |
| Parkinson’s disease | Rat | Liver allogeneic mitochondria conjugated with Pep-1 | Intranasal infusion | [63] |
| Alzheimer’s disease | Mouse | HeLa cell-derived mitochondria | Tail intravenous injection | [60][77] |
| Cognitively impaired aged mice | Mouse | Liver allogeneic mitochondria isolated from young mice | Injected into the hippocampus of aged mice | [78] |
| Diabetes-associated cognitively impaired mice | Mouse | Platelet-derived mitochondria | Intracerebroventricular injection | [79] |
| Chronic mild stressed—aged rats | Rat | Brain-derived mitochondria from young rats | Injected intracerebroventricularly in aged rats | [80] |
| Aging | Mouse | Liver-derived allogeneic mitochondria from young rats | Intravenous injection | [68] |
| CNS injury | Mouse | Cerebral cortex-derived allogeneic mitochondria whose DJ1 protein was modified with O-GlcNAcylation | Intraventricular injection | [81] |
| Traumatic brain injury | Mouse | Liver/muscle-derived autologous mitochondria | Injected into the cerebral cortex | [71] |
| Brain stroke | Mouse | Bone marrow mesenchymal stem cell-derived allogenic mitochondria | Intranasal administration | [82] |
| Schizophrenia | Rat | Rat brain-derived allogeneic mitochondria | Bilateral injection of into medial prefrontal cortex | [61][62] |
| Lipopolysaccharide (LPS)-induced model of depression | Mouse | Hippocampus-derived allogeneic mitochondria | Intravenous injection | [75] |
| Spinal cord injury | Rat | Soleus muscle-derived allogeneic mitochondria | Injected into spinal cord | [69] |
| Spinal cord ischemia | Rat | Soleus muscle-derived allogeneic mitochondria | Intravenous transplantation via the jugular vein | [83] |
| Spinal cord injury | Rat | Soleus muscle-derived allogenic mitochondria | Injection into spinal cord via intraparenchymal route | [70] |
| Neuromuscular limb injury | Mouse | NR | Systemic injection | [84] |
| Lower limb ischemia–reperfusion injury | Mouse | Human umbilical cord mesenchymal stem cell-derived mitochondria | Gastrocnemius muscle injection | [85] |
| Optic nerve injury | Rat | Liver-derived allogeneic mitochondria | Intravitreal injections | [86] |
| Mitochondria transplantation in humans | ||||
| Recurrent pregnancy failure cases | Human | Ovarian cortical tissue-derived autologous mitochondria | Mitochondria transferred to human oocytes, intracytoplasmic sperm injection | [66] |
| Ischemia–reperfusion-associated myocardial dysfunction | Human (infants) | Rectus abdominis muscle-derived autologous mitochondria | Injected into the myocardium affected by ischemia–reperfusion | [64] |
| Single large-scale mitochondrial DNA (mtDNA) de novo deletion syndromes (SLSMDs) | Human (infants) | Maternal PBMC-derived mitochondria transplanted into the patients’ enriched CD34+ cells after leukapheresis | Transfused into patients | [65] |
NR—not reported.
References
- Ricci, J.-E.; Muñoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the P75 Subunit of Complex I of the Electron Transport Chain. Cell 2004, 117, 773–786.
- Kim, J.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414.
- Diano, S.; Horvath, T.L. Mitochondrial Uncoupling Protein 2 (UCP2) in Glucose and Lipid Metabolism. Trends Mol. Med. 2012, 18, 52–58.
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics–Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463.
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of Mitochondrial Oxidative Phosphorylation through Cell Signaling. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1701–1720.
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469.
- Papa, S.; Rasmo, D.D.; Technikova-Dobrova, Z.; Panelli, D.; Signorile, A.; Scacco, S.; Petruzzella, V.; Papa, F.; Palmisano, G.; Gnoni, A.; et al. Respiratory Chain Complex I, a Main Regulatory Target of the cAMP/PKA Pathway Is Defective in Different Human Diseases. FEBS Lett. 2012, 586, 568–577.
- White, R.J.; Reynolds, I.J. Mitochondrial Depolarization in Glutamate-Stimulated Neurons: An Early Signal Specific to Excitotoxin Exposure. J. Neurosci. 1996, 16, 5688–5697.
- Basu, B.; Desai, R.; Balaji, J.; Chaerkady, R.; Sriram, V.; Maiti, S.; Panicker, M.M. Serotonin in Pre-Implantation Mouse Embryos Is Localized to the Mitochondria and Can Modulate Mitochondrial Potential. Reproduction 2008, 135, 657–669.
- Brenner-Lavie, H.; Klein, E.; Ben-Shachar, D. Mitochondrial Complex I as a Novel Target for Intraneuronal DA: Modulation of Respiration in Intact Cells. Biochem. Pharmacol. 2009, 78, 85–95.
- Blank, H.M.; Li, C.; Mueller, J.E.; Bogomolnaya, L.M.; Bryk, M.; Polymenis, M. An Increase in Mitochondrial DNA Promotes Nuclear DNA Replication in Yeast. PLoS Genet. 2008, 4, e1000047.
- Roubertoux, P.L.; Sluyter, F.; Carlier, M.; Marcet, B.; Maarouf-Veray, F.; Chérif, C.; Marican, C.; Arrechi, P.; Godin, F.; Jamon, M.; et al. Mitochondrial DNA Modifies Cognition in Interaction with the Nuclear Genome and Age in Mice. Nat. Genet. 2003, 35, 65–69.
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A.K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N.B.V. Lipid-Droplet Associated Mitochondria Promote Fatty-Acid Oxidation through a Distinct Bioenergetic Pattern in Male Wistar Rats. Nat. Commun. 2023, 14, 766.
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of Fatty Acid Degradation by Astrocytic Mitochondria Triggers Neuroinflammation and Neurodegeneration. Nat. Metab. 2023, 5, 445–465.
- Holper, L.; Ben-Shachar, D.; Mann, J. Multivariate Meta-Analyses of Mitochondrial Complex I and IV in Major Depressive Disorder, Bipolar Disorder, Schizophrenia, Alzheimer Disease, and Parkinson Disease. Neuropsychopharmacology 2019, 44, 837–849.
- Rollins, B.L.; Morgan, L.; Hjelm, B.E.; Sequeira, A.; Schatzberg, A.F.; Barchas, J.D.; Lee, F.S.; Myers, R.M.; Watson, S.J.; Akil, H.; et al. Mitochondrial Complex I Deficiency in Schizophrenia and Bipolar Disorder and Medication Influence. Complex Psychiatry 2017, 3, 157–169.
- Ni, P.; Ma, Y.; Chung, S. Mitochondrial Dysfunction in Psychiatric Disorders. Schizophr. Res. 2022; in press.
- Reddy, P.H. Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromol. Med. 2008, 10, 291–315.
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588.
- Dror, N.; Klein, E.; Karry, R.; Sheinkman, A.; Kirsh, Z.; Mazor, M.; Tzukerman, M.; Ben-Shachar, D. State-Dependent Alterations in Mitochondrial Complex I Activity in Platelets: A Potential Peripheral Marker for Schizophrenia. Mol. Psychiatry 2002, 7, 995–1001.
- Ben-Shachar, D.; Karry, R. Sp1 Expression Is Disrupted in Schizophrenia; A Possible Mechanism for the Abnormal Expression of Mitochondrial Complex I Genes, NDUFV1 and NDUFV2. PLoS ONE 2007, 2, e817.
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; et al. Mitochondrial Genes Are Altered in Blood Early in Alzheimer’s Disease. Neurobiol. Aging 2017, 53, 36–47.
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but Not Mitochondrial-encoded Oxidative Phosphorylation Genes Are Altered in Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. Alzheimer’s Dement. 2017, 13, 510–519.
- Zhu, Y.; Wang, Z.; Ni, J.; Zhang, Y.; Chen, M.; Cai, J.; Li, X.; Zhang, W.; Zhang, C. Genetic Variant in NDUFS1 Gene Is Associated with Schizophrenia and Negative Symptoms in Han Chinese. J. Hum. Genet. 2015, 60, 11–16.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102.
- Bubber, P.; Hartounian, V.; Gibson, G.E.; Blass, J.P. Abnormalities in the Tricarboxylic Acid (TCA) Cycle in the Brains of Schizophrenia Patients. Eur. Neuropsychopharmacol. 2011, 21, 254–260.
- Jia, D.; Wang, F.; Yu, H. Systemic Alterations of Tricarboxylic Acid Cycle Enzymes in Alzheimer’s Disease. Front. Neurosci. 2023, 17, 1206688.
- Bourgeron, T.; Chretien, D.; Poggi-Bach, J.; Doonan, S.; Rabier, D.; Letouzé, P.; Munnich, A.; Rötig, A.; Landrieu, P.; Rustin, P. Mutation of the Fumarase Gene in Two Siblings with Progressive Encephalopathy and Fumarase Deficiency. J. Clin. Investig. 1994, 93, 2514–2518.
- Guffon, N.; Lopez-Mediavilla, C.; Dumoulin, R.; Mousson, B.; Godinot, C.; Carrier, H.; Collombet, J.M.; Divry, P.; Mathieu, M.; Guibaud, P. 2-Ketoglutarate Dehydrogenase Deficiency, a Rare Cause of Primary Hyperlactataemia: Report of a New Case. J. Inherit. Metab. Dis. 1993, 16, 821–830.
- Stortz, J.A.; Hawkins, R.B.; Holden, D.C.; Raymond, S.L.; Wang, Z.; Brakenridge, S.C.; Cuschieri, J.; Moore, F.A.; Maier, R.V.; Moldawer, L.L.; et al. Cell-Free Nuclear, but Not Mitochondrial, DNA Concentrations Correlate with the Early Host Inflammatory Response after Severe Trauma. Sci. Rep. 2019, 9, 13648.
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular Mitochondrial DNA and Oxidatively Damaged DNA in Synovial Fluid of Patients with Rheumatoid Arthritis. Arthritis Res. Ther. 2003, 5, R234.
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces in Vivo and in Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000.
- Kageyama, Y.; Kasahara, T.; Kato, M.; Sakai, S.; Deguchi, Y.; Tani, M.; Kuroda, K.; Hattori, K.; Yoshida, S.; Goto, Y.; et al. The Relationship between Circulating Mitochondrial DNA and Inflammatory Cytokines in Patients with Major Depression. J. Affect. Disord. 2018, 233, 15–20.
- Ibrahim, M.; Strah, H.; Huang, H.; Krupnick, A.; Kreisel, D.; Hachem, R.; Trulock, E.; Alouch, A.; Gelman, A. Circulating Mitochondrial DNA Is Elevated in Patients with Immediate Primary Graft Dysfunction. J. Heart Lung Transplant. 2013, 32, S33.
- Varhaug, K.N.; Barro, C.; Bjørnevik, K.; Myhr, K.-M.; Torkildsen, Ø.; Wergeland, S.; Bindoff, L.A.; Kuhle, J.; Vedeler, C. Neurofilament Light Chain Predicts Disease Activity in Relapsing-Remitting MS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e422.
- Leurs, C.E.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Piehl, F.; Uitdehaag, B.M.; Killestein, J.; Van Horssen, J.; et al. Cerebrospinal Fluid mtDNA Concentration Is Elevated in Multiple Sclerosis Disease and Responds to Treatment. Mult. Scler. 2018, 24, 472–480.
- Kingwell, K. A Biomarker and a Target for Progressive MS. Nat. Rev. Drug Discov. 2009, 8, 771.
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. Ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064.
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin Inhibits Cytosolic Mitochondrial DNA–Induced Neuroinflammatory Signaling in Accelerated Aging and Neurodegeneration. J. Clin. Investig. 2021, 131, e150328.
- Tresse, E.; Marturia-Navarro, J.; Sew, W.Q.G.; Cisquella-Serra, M.; Jaberi, E.; Riera-Ponsati, L.; Fauerby, N.; Hu, E.; Kretz, O.; Aznar, S.; et al. Mitochondrial DNA Damage Triggers Spread of Parkinson’s Disease-like Pathology. Mol. Psychiatry, 2023; online ahead of print.
- Galizzi, G.; Di Carlo, M. Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Curr. Issues Mol. Biol. 2023, 45, 8586–8606.
- Daniels, T.E.; Zitkovsky, E.K.; Laumann, L.E.; Kunicki, Z.J.; Price, D.J.; Peterson, A.L.; Dennery, P.A.; Kao, H.-T.; Parade, S.H.; Price, L.H.; et al. Circulating Cell-Free Mitochondrial DNA and Depressive Symptoms Among Low-Active Adults Who Smoke. Psychosom. Med. 2024, 86, 37–43.
- Fernström, J.; Ohlsson, L.; Asp, M.; Lavant, E.; Holck, A.; Grudet, C.; Westrin, Å.; Lindqvist, D. Plasma Circulating Cell-Free Mitochondrial DNA in Depressive Disorders. PLoS ONE 2021, 16, e0259591.
- Ouyang, H.; Huang, M.; Xu, Y.; Yao, Q.; Wu, X.; Zhou, D. Reduced Cell-Free Mitochondrial DNA Levels Were Induced by Antipsychotics Treatment in First-Episode Patients with Schizophrenia. Front. Psychiatry 2021, 12, 652314.
- Melamud, M.M.; Buneva, V.N.; Ermakov, E.A. Circulating Cell-Free DNA Levels in Psychiatric Diseases: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 3402.
- Schulmann, A.; Ryu, E.; Goncalves, V.; Rollins, B.; Christiansen, M.; Frye, M.A.; Biernacka, J.; Vawter, M.P. Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Complex Psychiatry 2019, 5, 13–27.
- Shang, D.; Huang, M.; Wang, B.; Yan, X.; Wu, Z.; Zhang, X. mtDNA Maintenance and Alterations in the Pathogenesis of NeurodegenerativeDiseases. Curr. Neuropharmacol. 2023, 21, 578–598.
- Win, P.W.; Singh, S.M.; Castellani, C.A. Mitochondrial DNA Copy Number and Heteroplasmy in Monozygotic Twins Discordant for Schizophrenia. Twin Res. Hum. Genet. 2023, 26, 280–289.
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced Mitochondrial DNA Copy Number Is a Biomarker of Parkinson’s Disease. Neurobiol. Aging 2016, 38, e7–e216.
- Öğütlü, H.; Selçuk Esin, İ.; Bağış Erdem, H.; Tatar, A.; Burak Dursun, O. Mitochondrial DNA Copy Number Is Associated with Attention Deficit Hyperactivity Disorder. Psychiatr. Danub. 2020, 32, 168–175.
- Kumar, P.; Efstathopoulos, P.; Millischer, V.; Olsson, E.; Wei, Y.B.; Brüstle, O.; Schalling, M.; Villaescusa, J.C.; Ösby, U.; Lavebratt, C. Mitochondrial DNA Copy Number Is Associated with Psychosis Severity and Anti-Psychotic Treatment. Sci. Rep. 2018, 8, 12743.
- Suh, J.; Lee, Y.-S. Mitochondria as Secretory Organelles and Therapeutic Cargos. Exp. Mol. Med. 2024, 56, 66–85.
- Roushandeh, A.M.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial Transplantation as a Potential and Novel Master Key for Treatment of Various Incurable Diseases. Cytotechnology 2019, 71, 647–663.
- McCully, J.D.; Del Nido, P.J.; Emani, S.M. Mitochondrial Transplantation for Organ Rescue. Mitochondrion 2022, 64, 27–33.
- Wang, Z.-H.; Chen, L.; Li, W.; Chen, L.; Wang, Y.-P. Mitochondria Transfer and Transplantation in Human Health and Diseases. Mitochondrion 2022, 65, 80–87.
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of Isolated Mitochondria during Early Reperfusion for Cardioprotection. Am. J. Physiol.—Heart Circ. Physiol. 2009, 296, H94–H105.
- Shi, X.; Bai, H.; Zhao, M.; Li, X.; Sun, X.; Jiang, H.; Fu, A. Treatment of Acetaminophen-Induced Liver Injury with Exogenous Mitochondria in Mice. Transl. Res. 2018, 196, 31–41.
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; Li, Y.; Wang, Q. Muscle-Derived Autologous Mitochondrial Transplantation: A Novel Strategy for Treating Cerebral Ischemic Injury. Behav. Brain Res. 2019, 356, 322–331.
- Huang, P.-J.; Kuo, C.-C.; Lee, H.-C.; Shen, C.-I.; Cheng, F.-C.; Wu, S.-F.; Chang, J.-C.; Pan, H.-C.; Lin, S.-Z.; Liu, C.-S.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 2016, 25, 913–927.
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2019, 72, 587–604.
- Robicsek, O.; Ene, H.M.; Karry, R.; Ytzhaki, O.; Asor, E.; McPhie, D.; Cohen, B.M.; Ben-Yehuda, R.; Weiner, I.; Ben-Shachar, D. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr. Bull. 2018, 44, 432–442.
- Ene, H.M.; Karry, R.; Farfara, D.; Ben-Shachar, D. Mitochondria Play an Essential Role in the Trajectory of Adolescent Neurodevelopment and Behavior in Adulthood: Evidence from a Schizophrenia Rat Model. Mol. Psychiatry 2023, 28, 1170–1181.
- Chang, J.-C.; Chao, Y.-C.; Chang, H.-S.; Wu, Y.-L.; Chang, H.-J.; Lin, Y.-S.; Cheng, W.-L.; Lin, T.-T.; Liu, C.-S. Intranasal Delivery of Mitochondria for Treatment of Parkinson’s Disease Model Rats Lesioned with 6-Hydroxydopamine. Sci. Rep. 2021, 11, 10597.
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289.
- Jacoby, E.; Bar-Yosef, O.; Gruber, N.; Lahav, E.; Varda-Bloom, N.; Bolkier, Y.; Bar, D.; Blumkin, M.B.-Y.; Barak, S.; Eisenstein, E.; et al. Mitochondrial Augmentation of Hematopoietic Stem Cells in Children with Single Large-Scale Mitochondrial DNA Deletion Syndromes. Sci. Transl. Med. 2022, 14, eabo3724.
- Morimoto, Y.; Gamage, U.S.K.; Yamochi, T.; Saeki, N.; Morimoto, N.; Yamanaka, M.; Koike, A.; Miyamoto, Y.; Tanaka, K.; Fukuda, A.; et al. Mitochondrial Transfer into Human Oocytes Improved Embryo Quality and Clinical Outcomes in Recurrent Pregnancy Failure Cases. Int. J. Mol. Sci. 2023, 24, 2738.
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous Administration of Mitochondria for Treating Experimental Parkinson’s Disease. Mitochondrion 2017, 34, 91–100.
- Zhao, Z.; Yu, Z.; Hou, Y.; Zhang, L.; Fu, A. Improvement of Cognitive and Motor Performance with Mitotherapy in Aged Mice. Int. J. Biol. Sci. 2020, 16, 849–858.
- Gollihue, J.L.; Patel, S.P.; Eldahan, K.C.; Cox, D.H.; Donahue, R.R.; Taylor, B.K.; Sullivan, P.G.; Rabchevsky, A.G. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J. Neurotrauma 2018, 35, 1800–1818.
- Lin, M.-W.; Fang, S.-Y.; Hsu, J.-Y.C.; Huang, C.-Y.; Lee, P.-H.; Huang, C.-C.; Chen, H.-F.; Lam, C.-F.; Lee, J.-S. Mitochondrial Transplantation Attenuates Neural Damage and Improves Locomotor Function After Traumatic Spinal Cord Injury in Rats. Front. Neurosci. 2022, 16, 800883.
- Zhao, J.; Qu, D.; Xi, Z.; Huan, Y.; Zhang, K.; Yu, C.; Yang, D.; Kang, J.; Lin, W.; Wu, S.; et al. Mitochondria Transplantation Protects Traumatic Brain Injury via Promoting Neuronal Survival and Astrocytic BDNF. Transl. Res. 2021, 235, 102–114.
- Ben-Shachar, D. Mitochondrial Multifaceted Dysfunction in Schizophrenia; Complex I as a Possible Pathological Target. Schizophr. Res. 2017, 187, 3–10.
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial Dysfunction and Psychiatric Disorders. Neurochem. Res. 2009, 34, 1021–1029.
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801.
- Wang, Y.; Ni, J.; Gao, C.; Xie, L.; Zhai, L.; Cui, G.; Yin, X. Mitochondrial Transplantation Attenuates Lipopolysaccharide- Induced Depression-like Behaviors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 93, 240–249.
- Chang, J.-C.; Wu, S.-L.; Liu, K.-H.; Chen, Y.-H.; Chuang, C.-S.; Cheng, F.-C.; Su, H.-L.; Wei, Y.-H.; Kuo, S.-J.; Liu, C.-S. Allogeneic/Xenogeneic Transplantation of Peptide-Labeled Mitochondria in Parkinson’s Disease: Restoration of Mitochondria Functions and Attenuation of 6-Hydroxydopamine–Induced Neurotoxicity. Transl. Res. 2016, 170, 40–56.e3.
- Sweetat, S.; Nitzan, K.; Suissa, N.; Haimovich, Y.; Lichtenstein, M.; Zabit, S.; Benhamron, S.; Akarieh, K.; Mishra, K.; Barasch, D.; et al. The Beneficial Effect of Mitochondrial Transfer Therapy in 5XFAD Mice via Liver–Serum–Brain Response. Cells 2023, 12, 1006.
- Zhang, Z.; Wei, D.; Li, Z.; Guo, H.; Wu, Y.; Feng, J. Hippocampal Mitochondrial Transplantation Alleviates Age-Associated Cognitive Decline via Enhancing Wnt Signaling and Neurogenesis. Comput. Intell. Neurosci. 2022, 2022, 9325302.
- Ma, H.; Jiang, T.; Tang, W.; Ma, Z.; Pu, K.; Xu, F.; Chang, H.; Zhao, G.; Gao, W.; Li, Y.; et al. Transplantation of Platelet-Derived Mitochondria Alleviates Cognitive Impairment and Mitochondrial Dysfunction in Db/Db Mice. Clin. Sci. 2020, 134, 2161–2175.
- Javani, G.; Babri, S.; Farajdokht, F.; Ghaffari-Nasab, A.; Mohaddes, G. Mitochondrial Transplantation Improves Anxiety- and Depression-like Behaviors in Aged Stress-Exposed Rats. Mech. Ageing Dev. 2022, 202, 111632.
- Park, J.H.; Tanaka, M.; Nakano, T.; Licastro, E.; Nakamura, Y.; Li, W.; Esposito, E.; Mandeville, E.T.; Chou, S.H.-Y.; Ning, M.; et al. O-GlcNAcylation Is Essential for Therapeutic Mitochondrial Transplantation. Commun. Med. 2023, 3, 169.
- Hosseini, L.; Karimipour, M.; Seyedaghamiri, F.; Abolhasanpour, N.; Sadigh-Eteghad, S.; Mahmoudi, J.; Farhoudi, M. Intranasal Administration of Mitochondria Alleviated Cognitive Impairments and Mitochondrial Dysfunction in the Photothrombotic Model of mPFC Stroke in Mice. J. Stroke Cerebrovasc. Dis. 2022, 31, 106801.
- Fang, S.-Y.; Roan, J.-N.; Lee, J.-S.; Chiu, M.-H.; Lin, M.-W.; Liu, C.-C.; Lam, C.-F. Transplantation of Viable Mitochondria Attenuates Neurologic Injury after Spinal Cord Ischemia. J. Thorac. Cardiovasc. Surg. 2021, 161, e337–e347.
- Alway, S.E.; Paez, H.G.; Pitzer, C.R.; Ferrandi, P.J.; Khan, M.M.; Mohamed, J.S.; Carson, J.A.; Deschenes, M.R. Mitochondria Transplant Therapy Improves Regeneration and Restoration of Injured Skeletal Muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 493–507.
- Zeng, J.; Liu, J.; Ni, H.; Zhang, L.; Wang, J.; Li, Y.; Jiang, W.; Wu, Z.; Zhou, M. Mitochondrial Transplantation Reduces Lower Limb Ischemia-Reperfusion Injury by Increasing Skeletal Muscle Energy and Adipocyte Browning. Mol. Ther.—Methods Clin. Dev. 2023, 31, 101152.
- Nascimento-dos-Santos, G.; de-Souza-Ferreira, E.; Lani, R.; Faria, C.C.; Araújo, V.G.; Teixeira-Pinheiro, L.C.; Vasconcelos, T.; Gonçalo, T.; Santiago, M.F.; Linden, R.; et al. Neuroprotection from Optic Nerve Injury and Modulation of Oxidative Metabolism by Transplantation of Active Mitochondria to the Retina. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165686.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
996
Revisions:
3 times
(View History)
Update Date:
08 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No