Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yan, H.; He, L.; Lv, D.; Yang, J.; Yuan, Z. Dysregulated JNK Signaling Pathway in Human Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/55970 (accessed on 27 July 2026).
Yan H, He L, Lv D, Yang J, Yuan Z. Dysregulated JNK Signaling Pathway in Human Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/55970. Accessed July 27, 2026.
Yan, Huaying, Lanfang He, De Lv, Jun Yang, Zhu Yuan. "Dysregulated JNK Signaling Pathway in Human Diseases" Encyclopedia, https://encyclopedia.pub/entry/55970 (accessed July 27, 2026).
Yan, H., He, L., Lv, D., Yang, J., & Yuan, Z. (2024, March 07). Dysregulated JNK Signaling Pathway in Human Diseases. In Encyclopedia. https://encyclopedia.pub/entry/55970
Yan, Huaying, et al. "Dysregulated JNK Signaling Pathway in Human Diseases." Encyclopedia. Web. 07 March, 2024.
Copy Citation
JNK is named after c-Jun N-terminal kinase, as it is responsible for phosphorylating c-Jun. As a member of the mitogen-activated protein kinase (MAPK) family, JNK is also known as stress-activated kinase (SAPK) because it can be activated by extracellular stresses including growth factor, UV irradiation, and virus infection. Functionally, JNK regulates various cell behaviors such as cell differentiation, proliferation, survival, and metabolic reprogramming. Dysregulated JNK signaling contributes to several types of human diseases.
JNK signaling pathways
metabolic disorders
neurological diseases
chronic inflammatory diseases
cancer
infectious diseases
JNK inhibitors
1.Introduction
As discussed above, JNK activity is strictly regulated within cells. Once the JNK pathway is dysregulated, it is harmful to cells and ultimately aggravates disease conditions. Dysregulated JNK signaling contributes to a variety of diseases involving metabolic disorders, chronic inflammation, autoimmune diseases, neurodegeneration, cancer, infectious diseases, and other diseases such as ischemia/reperfusion injury, hearing loss/deafness, and kidney fibrosis.
2. The Role of the Dysregulated JNK Pathway in Metabolic Disorders
Metabolic syndrome, a metabolic disorder-associated disease, mainly includes obesity, type 2 diabetes (T2D), cardiovascular disorders, and non-alcoholic steatohepatitis (NASH). The dysregulated JNK pathway plays a role in the onset and progression of metabolic syndromes.
2.1. The JNK Pathway in Obesity
It is well known that obesity is caused by excessive fat accumulation [1][2]. Obesity often leads to several health problems, such as insulin resistance and T2D [3], fatty liver disease, cardiovascular disease, and cancer [4]. Currently, obesity-driven diabetes is becoming a global metabolic disorder [5]. Although the mechanisms for obesity-triggering T2D are not fully understood, inflammatory kinases play a role in insulin resistance [1]. As an inflammatory kinase, JNK is activated by pro-inflammatory cytokines and mediates the transition from obesity to T2D [1].
Previous studies have shown that obesity is a chronic inflammatory disease [1][6]. The evidence is as follows: a large number of macrophages are observed in the visceral adipose tissue. Moreover, macrophages in adipose tissues also secrete pro-inflammatory cytokines, including TNFα and IL-1, which then activate JNK kinases [6]. In addition, JNKs are also stimulated by high-fat diets and are activated by TNFα and IL-1 [5][7][8]. Obesity is also associated with the increase in adipocyte mass, adipokine secretion, and free fatty acids (FFAs), all of which can activate JNK kinases [9]. For instance, FFAs cause JNK activation in cultured 3T3-L1 cells [10]. In adipose tissue, FFAs activate JNK by promoting the activation of the Src–MLK axis [11][12]. Additionally, the scaffold protein JIP1 has been shown to link the Src–MLK-JNK axis with the FFA–JNK signaling axis [11][12]. Importantly, JIP−/− and MLK−/− mice were protected from obesity, showing that the JIP1-mediated Src–MLK signaling axis is needed for obesity [11][13][14][15]. Overall, these findings suggest that the JNK pathway plays a critical role in obesity.
2.2. The JNK Pathway in Insulin Resistance and T2D
High-fat diets (HFDs) trigger JNK over-activation, which leads to insulin resistance [16]. The opinion that JNK functions in insulin resistance is supported by the following observations: (1) JNK over-activation is observed during obesity, and mice with a JNK1 deficiency display a significantly improved insulin sensitivity level [7][16]; (2) JNK1 knockout mice are protected from obesity [16][17]; (3) mice lacking JNK2 also display an improved insulin sensitive phenotype [17]; and (4) there are some mutations in the JIP1 coding gene (JNK’s scaffold) in patients with type II diabetes [18].
As for the action mechanism, Solinas et al. provided four distinct mechanisms to explain the involvement of JNK in T2D: (1) the phosphorylation of IRS1/2; (2) contribution to metabolic inflammation; (3) negative regulation of the TSH–thyroid hormone axis; and (4) inhibition of PPARa-FGF21 signaling [16]. These four mechanisms are summarized in Figure 1. The first mechanism is supported by the report that anti-TNFα treatment ameliorates insulin sensitivity [19] and also by the findings that TNFα-activated JNK directly phosphorylates IRS1/2, causing insulin resistance [16][20]. Specifically, Tanti et al. found that phosphorylation of IRS1 by JNK inhibits its interaction with insulin receptor (IR), causes defective IRS1 tyrosine phosphorylation, and importantly, abrogates IRS downstream PI3K-AKT signaling, thus promoting FOXO1-mediated gluconeogenesis, and finally causing insulin resistance (Figure 1A) [21][22][23]. Notably, both JNK1 and JNK2 can phosphorylate IRS1 [17], and JNK is also able to phosphorylate IRS2 [22][24][25].
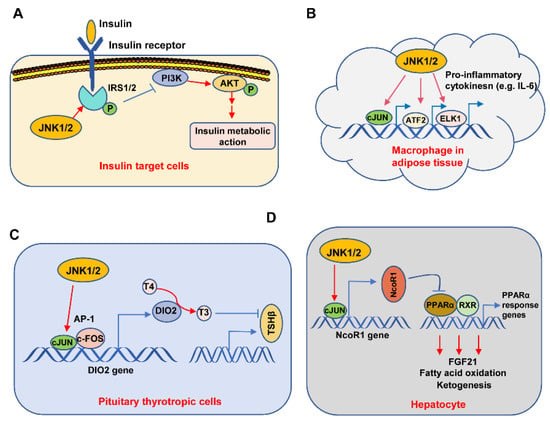
Figure 1. Molecular mechanisms for the involvement of the JNK pathway in insulin resistance. The JNK1/2 kinases were important for obesity-driven insulin resistance through four mechanisms: (A) In insulin-target cells, JNK1/2 directly phosphorylates IRS1 and IRS2 at serine and threonine residues, resulting in reduced tyrosine phosphorylation of IRS1/2 molecules, which further weakens the response of the PI3K/AKT signaling pathway to insulin. (B) In adipose tissue macrophages, JNK1/2 phosphorylates some transcriptional factors such as c-JUN, ATF2, and ELK1, stimulating gene transcription of pro-inflammatory cytokines, leading to increased levels of inflammatory “M1” cytokines (e.g., IL-6), which ultimately drive insulin resistance. (C) In pituitary thyrotropic cells, JNK1/2 phosphorylates c-JUN and c-FOS, sustaining the transcription and subsequent expression of DIO2, which mediates the shift from T4 to T3, finally leading to a T3-dependent reduction of TSHβ production. The reduced TSH ultimately causes low circulating levels of thyroid hormone, increases metabolic efficiency, and increases adiposity, finally driving insulin resistance. (D) In hepatocytes, JNK1/2 phosphorylates c-JUN, sustaining the transcription and expression of the transcription co-repressor NcoR1. Then, NcoR1 represses PPARα-driven gene expression, causing a reduction in FGF21 production, fatty acid oxidation, and ketogenesis, ultimately promoting fatty liver and insulin resistance.
Han et al. found that JNK activity in macrophages of adipose tissue contributes to obesity-induced insulin resistance and inflammation [26]. Mechanistically, the activated JNK1/2 promotes c-Jun, ATF-2, and ELK1 to bind to the promoters of pro-inflammatory cytokine-coding genes such as IL-6, subsequently causing the transcription of these genes, finally resulting in the secretion of pro-inflammatory cytokines from macrophages. Similarly, Perry et al. found that JNK signaling promotes the secretion of IL-6, causing an increase in FFAs that further facilitate the liver’s ability to synthesize glucose, finally resulting in insulin resistance [27]. Together, these studies indicate that JNK activation exacerbates the progression of T2D through the promotion of metabolic inflammation (Figure 1B). In regard to the regulation of the TSH–thyroid hormone axis by JNK, Belgardt et al. discovered that the expression levels of T3 and TSHβ in the pituitary were increased in HFD-feeding mice, whose JNK1 is deficient in the central nervous system, indicating that JNK inhibits the TSH–thyroid hormone axis. Moreover, body mass and liver triglyceride content were also reduced in these mice [28]. Similarly, Sabio et al. found that JNK1 deficiency in the CNS also caused a decrease in adiposity in HFD-feeding mice. Taken together, these studies showed that HFD-stimulated JNK1 promotes adiposity by negatively regulating the TSH–thyroid axis [29]. Moreover, the activated JNK promotes AP-1 transcription factors, which include c-Jun and c-Fos, to promote Dio2 gene transcription and expression. Then, Dio2 makes T4 convert into T3, which further inhibits TSHβ gene expression (Figure 1C) [30]. Subsequently, the reduced TSHβ results in a reduction of thyroid hormone and an increase in adiposity, which finally leads to insulin resistance. Notably, Vernia et al. found that HFD-feeding-activated JNK3 in hypothalamic neurons is needed for controlling appetite for food [31]. Interestingly, in contrast to control animals, HFD-feeding JNK3 knockout mice are apt to develop obesity and subsequent insulin resistance, suggesting neuron-specific JNK3 plays a role in improving insulin resistance [31].
Vernia et al. found that hepatocyte-specific JNK1/2 knock-out improved insulin sensitivity and significantly ameliorated fatty liver in a FGF21-dependent manner [32][33]. Mechanistically, JNK first stimulates c-JUN to bind to the promoter of the NcoR1 gene and promotes the transcription and subsequent expression of NCOR1. As a transcriptional corepressor, NCOR1 further represses the PPARα and RXR-mediated PPARα response genes, such as FGF21 expression, thus affecting fatty acid oxidation and ketogenesis, which finally result in insulin resistance (Figure 1D). In addition to these above mechanisms, JNK activity can affect insulin secretion from pancreatic β-cells. In the pancreatic β-cell, the elevated FFAs in plasma during obesity will result in the sustained activation of JNK, which, in turn, negatively regulates insulin secretion and finally causes β-cell dysfunction and death [1][34]. Collectively, these studies demonstrate that JNK plays an important role in obesity and type II diabetes.
2.3. JNK in Atherosclerosis
Atherosclerosis is a multi-step systemic inflammation and metabolic disease, which ultimately leads to myocardial infarction or stroke [6]. Both obesity and diabetes are known to contribute to atherosclerosis. Atherosclerosis onset and progression involve the following events: (1) An initiating stimulus, including vascular injury, hypercholesteremia, or chronic inflammation. These stimuli cause endothelial cell dysfunction and/or apoptosis, thus increasing vessel wall permeability to lipids and local inflammation. Under these conditions, endothelial cells then express cytokines that recruit monocytes and leukocytes to the area; (2) monocytes then trans-migrate across the blood vessel wall and differentiate into macrophages. As the vessel is lipid-laden, the macrophages ingest the low-density lipoproteins (LDLs), and then turn into foam cells. These foam cells further promote lesion progression by increasing local inflammation and ROS production; (3) under the actions of endothelial cell-secreted cytokines, T and B lymphocytes are recruited to the plaque. Smooth muscle cells are also recruited to the lumen and begin to proliferate and secrete collagen, elastin, and other extracellular matrix proteins [6]. Although multiple cell types contribute to this complex process, elevated JNK activity is observed in each of these steps [6]. As mentioned above, JNK1 or JNK2 plays an important role in inflammation and cytokine production. Moreover, JNK1 or JNK2 is expressed in all of the cell types relevant to the onset and progression of atherosclerosis: endothelial cells, smooth muscle cells, macrophages, and T cells, all of which are associated with the formation and development of atherosclerosis. Specifically, JNK1 promotes apoptosis in the endothelium after chronic inflammation and promotes atherosclerosis. Similar to the endothelium, JNK1 in bone marrow-derived immune cells (including monocytes) also promotes apoptosis after chronic inflammation, which leads to less atherosclerosis in mice. JNK2 knockout mice show less atherosclerosis through a reduced number of foam cell formation. These studies suggest that JNK is involved in inflammation and cytokine production, mediates the apoptosis of the endothelium, immune cells, and foam cells, and has different functions in different cells (macrophages, T cells, and endothelial cells). Previous studies have shown that JNK over-activation promotes the progression of atherosclerosis [6][35]. Furthermore, Ricci et al. found that JNK2 ablation can mitigate atherosclerosis progression, implying that JNK2 can promote atherosclerotic lesion formation [36]. However, Babaev et al. demonstrated that a hematopoietic cell-specific JNK1 deficiency promoted atherosclerosis in LDLR−/− mice. By contrast, Nofer et al. found that JNK promoted atherosclerosis onset. Similarly, Amini et al. found that JNK1 ablation protected atherosclerosis formation [37]. These studies suggest that JNK is critical for the onset and progression of atherosclerosis.
2.4. JNK in Non-alcoholic Fatty Liver Disease (NAFLD)
NAFLD includes non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis (NASH) [38]. Hypertension and cardiovascular diseases, hepatic gluconeogenesis-triggered hyperglycemia, and insulin resistance-caused hyperinsulinemia all lead to NAFL disease [38]. Although the mechanism is not fully understood, JNK activation in response to hepatic metabolic stresses, including inflammation, cytokines, de novo lipogenesis, and lipolysis, has been identified as a reason for NAFL/NASH [39][40][41][42][43].
It is well known that the liver contains about 60% parenchymal cells and over 30% non-parenchymal cells, including hepatic stellate cells. Among these cells, hepatocytes, a type of parenchymal cell, are the cornerstone of the liver. Hepatocytes express three JNK isoforms [44]. Diets, especially hyper-nutrition-type diets, including HFD, easily activate hepatic JNK [33]. Mechanically, HFD first activates JNK in the hepatocytes [45][46]. Then, the activated p-JNK further phosphorylates SAB, a mitochondrial outer membrane protein, resulting in mitochondrial ROS production, which further activates the ASK1-MKK4/7-JNK axis [44][47], thus forming a JNK-SAB-ROS-ASK1-JNK feedback loop, finally resulting in sustained JNK activation. This feedback loop is considered a critical mechanism in the development of hepatic steatosis [27][48]. The important role of JNK in NASH is proved by the following reports: ASK1 knockout mice are protected from hepatic steatosis [38][49]; MLK knockout mice also reduce the level of triglycerides [50]; similarly, JNK knockout mice avoid HFD-triggered NASH [51].
3. The Role of JNK Signaling in Neurological Diseases
JNK signaling exerts different roles in neurogenesis, axonal growth, axonal transport, and brain metabolism [52]. The deregulated JNK pathway leads to developmental axonal defects. Moreover, the over-activation of the JNK pathway, which means the kinase activity of JNK is constitutively increased, also contributes to CNS pathologies and motor neuron (MN) diseases [53]. Here, the researchers briefly summarize the function of JNK in CNS pathologies such as glioblastoma progression, neurodegenerative diseases, and CNS regeneration/repair after an injury.
3.1. The JNK Pathway in Glioblastoma Progression
The JNK pathway is highly active in the central nervous system (CNS) as compared with other tissues. Previous studies revealed a dual role of JNK in cell death and survival, which is very important for glioblastoma (GB) tumorigenesis and neurodegeneration [54][55][56]. GB is a malignant brain tumor [52][57]. Over-activation of the JNK pathway is a hallmark related to glial cell proliferation and cancer stem cell-like properties [56][58][59]. Recent findings showed that JNK activity correlates with GB aggressiveness and that JNK promotes GB progression and infiltration [56][60]. Mechanically, JNK activation leads to upregulation of matrix metalloproteases (MMPs) [56], causing extracellular matrix degradation and promoting GB cell infiltration [56]. Interestingly, the progressive increase of JNK pathway activation in GB samples is associated with the localization of the Grnd receptor’s ligand in glial cells [60]. Recently, Jarabo et al. found that GB cells can secrete Impl2 to induce neuronal changes, contributing to tumor progression. Importantly, this process is also regulated by JNK activity [61]. Together, these studies indicate that GB cells use different ways to activate the JNK signaling pathway, further promoting GB progression.
3.2. The JNK Pathway in Neurodegenerative Diseases
Parkinson’s disease (PD), Alzheimer’s disease (AD), and Huntington’s disease (HD) are the three most common neurodegenerative diseases [5]. CNS-specific JNK3 has been shown to be a mediator for these neurodegenerative diseases. In AD patients, there are many diffuse plaques in cortical and hippocampal neurons [5][62]. These plaques mainly contain β-amyloid peptide (Aβ) and hyperphosphorylated Tau protein. Aβ induces neuronal apoptosis through JNK-dependent downregulation of Bcl-w, resulting in cortical neuron apoptosis [63]. JNK is also responsible for phosphorylating Tau to promote tangle formation [64]. In addition, JNK3 over-activation promotes AD progression as JNK3-deficient mice display synapse regrowth and a decrease in Aβ [52][65]. These mice are protected from Aβ-induced neuronal cell apoptosis through a JNK3-mediated AP-1-dependent FasL manner [62][63][66].
JNK also mediates PD progression. Choi et al. found that neurotoxin-induced JNK3 activation mediated dopaminergic neuron death in the substantia nigra [67]. JNK over-activation induces apoptosis of dopaminergic neurons [68]. Consistently, JNK3−/− and JNK2−/− mice prevented dopaminergic cell death induced by the neurotoxin MPTP [5]. Mechanistically, JNK-mediated autophagy and apoptosis play key roles in PD progression [68]. Bcl-2 has been identified as a critical protein with the ability to suppress autophagy and apoptosis through inhibiting Beclin-1 and Bax, respectively. Moreover, both JNK and P38 mediate dopaminergic neuron death in PD via apoptosis by increasing the Bax/Bcl-2 ratio. With regard to autophagy, JNK-mediated BCL-2 phosphorylation also suppresses the functions of Bcl-2 in autophagy. Additionally, recent findings indicate that receptor-interacting protein kinase 1 (RIPK1) is upregulated in PD in in vitro and in vivo models. RIPK1 promotes cell apoptosis and reactive oxygen species (ROS) production through the activation of the JNK pathway, thus leading to dopaminergic cell death. These discoveries suggest that JNK, together with other factors such as p38, BCL-2, Bax, Beclin-1, and RIPK1, mediate the progression of PD.
HD disease is caused by the degeneration of projection neurons in the striatum and the cerebral cortex. In HD patients, there are some amplifications of glutamine repeat (poly Q) in huntingtin protein (HTT), which trigger protein aggregation and neuronal degeneration [69]. During this process, polyQ-containing proteins activate JNKs. Furthermore, Morfini et al. demonstrated that aberrant HTT would impair axonal transport (FAT) through the JNK-mediated phosphorylation of the kinesin-1 protein [70]. Furthermore, JNK kinase inhibitors conferred neuroprotection in an HD mouse model [71], providing further evidence that JNK functions in HD progression. Taken together, these observations have demonstrated that the JNK signaling pathway is critical for the progress of neurodegenerative diseases.
3.3. JNK in Excitatory Toxicity of Hippocampal Neurons
The expression of JNK3 in the fore- and hind-brain regions is high. Normally, mice treated with kainic acid, a neurotoxic reagent, display significant apoptosis of hippocampal neurons [5]. JNK3 knockout mice also avoid glutamate-induced excitatory toxicity. Similarly, mice knocking in a mutant c-Jun (S63A and S73A) that cannot be phosphorylated by JNK display resistance against glutamate-induced neuronal toxicity [72]. These studies indicate that JNK3 activity promotes neuronal excitatory toxicity.
3.4. Abnormal Activation of the JNK Pathway in Multiple Sclerosis
Multiple sclerosis (MS) is a chronic demyelinating neurodegenerative disease of the CNS, manifesting as myelin sheath damage. Zhang et al. found that JNK was overactivated in chronic active MS plaques [73]. Bagnoud et al. also found that the phosphorylated JNK is increased during demyelination-triggered MS [74]. During the progression of MS, the phosphorylation of JNK by ASK1 plays critical roles in multiple processes, such as oligodendrocyte destruction, demyelination, neuroinflammation, immune dysregulation through T cell activation (T cell apoptosis), and oxidative damage. For instance, the TLR-ASK1-JNK pathway is active in glial cells and important for autoimmune demyelinating disorders [75]. In addition to the TLR (Toll-like receptor), other factors, including reactive oxygen species (ROS), oxidative stress, and inflammation, can activate ASK1 during MS progression [76]. These reports indicate that the over-activated ASK1-MKK4/7-JNK signaling axis plays a critical role in the pathogenesis of MS; that is, deregulated JNK signaling contributes to MS progression [73][74][76].
3.5. JNKs in CNS Damage Repair
Some factors, such as brain stroke and spinal cord injury, often trigger CNS damage. Because the self-repair or regenerative ability of this tissue is limited, the damaged CNS often displays permanent disabilities [52]. Previous studies have shown that JNK signaling has an important function in degeneration and the repair of nerve injuries. That is, retrograde transport of JNK in the injured axons changes the transcription of ATF3, thus affecting axon growth [77][78]. In addition, suppression of JNK signaling delays axonal degeneration [79]. DLK-JNK signaling also contributes to axonal regeneration after spinal cord injury [80][81][82][83]. Additionally, JNK3 interacts with KLF9 to inhibit axon regeneration upon nerve injury [84]. Overall, these reports clearly show the relevance of JNK to regeneration/repair after CNS injury.
4. JNK Pathway in Chronic Inflammatory Disease
Autoimmune disorders are a group of chronic inflammatory diseases, such as autoimmune arthritis (AA), osteoarthritis (OA), and inflammatory bowel disease (IBD). Here, the researchers take AA, OA, and IBD as examples to demonstrate the function of JNK in chronic inflammatory disease.
4.1. The JNK Pathway in Autoimmune Arthritis
Rheumatoid arthritis (RA), psoriatic arthritis (PsA), and ankylosing spondylitis (AS) are three common autoimmune arthritis (AA) diseases [85]. The common characteristics of these diseases is the destruction of the joints and bones. Interestingly, extensive lymphocyte infiltration occurs in the inflamed joints in AA [85]. Also, pro-inflammatory cytokines, for instance, tumor necrosis factor-alpha (TNF-α), are significantly elevated in serum and joint fluid [85][86].
Fukushima et al. found that the phosphorylation of JNK was increased in the joint tissues of mice with inflammatory arthritis [87][88]. Similarly, JNK activation was also observed in synovial tissues from patients with psoriatic arthropathy and RA patients [89][90]. Recently, Li et al. found that JNK signaling was also involved in the pathogenesis of ankylosing spondylitis [91]. Mechanically, JNK is activated by pro-inflammatory cytokines, including TNF-α, causing autoimmune arthritis [85]. Additionally, JNK also regulates the expression of metalloproteases, promoting joint destruction [5], as JNK1/JNK2-deficient rheumatoid arthritis mice were protected from joint damage and reduced the expression level of metalloproteinase [5]. These studies clearly suggested that deregulated JNK signaling plays a role in autoimmune arthritis.
4.2. The JNK Pathway in Osteoarthritis
Osteoarthritis (OA) is a joint degeneration-related disease. The characteristics of OA mainly include several pathologic changes, such as joint inflammation, erosion of articular cartilage, and osteophyte formation [92]. JNK has been shown to aggravate this pathological process. For example, upon activation, JNK promotes cartilage destruction. Specifically, JNK is first activated by the pro-inflammatory cytokine TNFα; then, the activated JNK phosphorylates c-Jun, promoting MMP-13 (a cartilage-degrading enzyme) transcription and expression, which finally causes a decrease in proteoglycan synthesis. Additionally, excessive MMP-13 expression in chondrocytes contributes to cartilage degeneration [5][93]. Additionally, Yang et al. found that the elevated levels of CXCL8 and CXCL11 in the synovial fluids also aggravate OA progression by activating the JNK pathway, which mediates the apoptosis of chondrocytes [94].
4.3. JNK Functions in Chronic Inflammatory Bowel Disease
Crohn’s disease and ulcerative colitis are two chronic inflammatory bowel diseases (IBDs). Several pro-inflammatory cytokines in the intestinal microenvironments, for example, TNFα, can activate JNK signaling, which accelerates the development of IBD [95]. Previous studies showed that JNK was highly activated in the intestinal tissues from IBD patients [95][96]. Similarly, Mistsuyama et al. found that the active form of JNK was expressed in the nuclei of intestinal cells, macrophages, and lymphocytes, further providing indirect evidence that pro-inflammatory cytokines activate the JNK pathway in IBD [96]. Chromik et al. investigated the function of JNK isoforms in IBD and found that JNK2 deficiency aggravated DSS-induced colitis in mice; however, JNK1 deficiency did not completely block colitis development [97]. By contrast, the JNK inhibitor SP600125 partially protected against colonic injury [95].
5. Pro- and Anti-Oncogenic Roles of JNK in Cancer
The JNK pathway has a dual role in different types of cancers, including hepatocellular carcinoma (HCC), pancreatic cancer, prostate cancer, multiple myeloma and oral cancer, non-small-cell lung cancer (NSCLC), glioblastoma, papilloma, intestinal tumors, breast cancer, squamous cell carcinoma, Burkitt’s lymphoma, and ovarian cancer [5][52][98][99][100][101]. The dual effects of JNK in oncogenesis are summarized in Figure 2. On the one hand, JNK can promote cell death, eliminating pre-tumorigenic cells. On the other hand, it can also stimulate tumorigenesis [5][54].
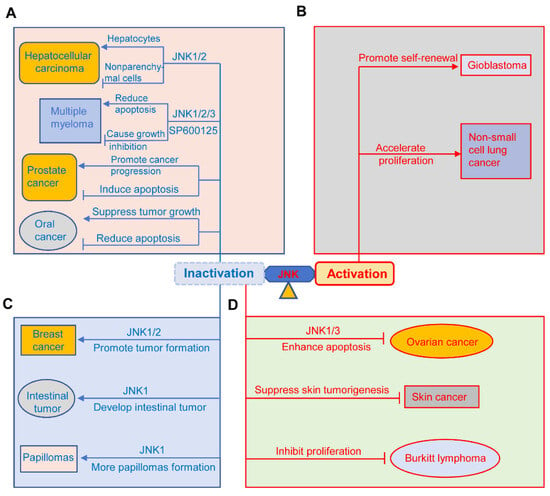
Figure 2. Pro-oncogenic and anti-oncogenic roles of JNK signaling in cancers. (A) The JNK signaling plays a dual role in the occurrence and progression of different kinds of cancer, including hepatocellular carcinoma, multiple myeloma, prostate cancer, and oral cancer. (B) JNK signaling promotes the progression of glioblastoma and non-small-cell lung cancer. (C) Deficiency or inhibition of different JNKs promotes tumor formation in breast cancer, intestinal tumors, and papilloma. (D) Inhibition of the JNK pathway suppresses the tumorigenesis of ovarian cancer, skin cancer, and Burkitt lymphoma. Activation indicates sustained activation of the JNK pathway; inactivation represents deficiency of JNK1, JNK2, JNK3, or compound deficiency of JNK1 and JNK2; inactivation also indicates inhibition of JNK activity by JNK inhibitors such as SP600125.
5.1. The Dual Role of JNK in Hepatocellular Carcinoma (HCC), Multiple Myeloma, Prostate Cancer, and Oral Cancer
JNK has a tumor promoting or suppressing function in HCC, pancreatic cancer, multiple myeloma, and oral cancer (Figure 2A). JNK1 deficiency in hepatocytes and JNK2 deficiency in the liver promote HCC progress, indicating its role in inhibiting tumor development [101]. Similarly, JNK1 deficiency significantly decreases susceptibility to diethylnitrosamine-induced hepatocarcinogenesis [5]. However, JNK has an oncogenesis role in non-parenchymal cells of the liver through the production of pro-tumorigenic cytokines [101]. Hideshima et al. found that the JNK inhibitor SP600125 arrests MM cells in the cell cycle and causes cell growth inhibition through activation of the NF-κB pathway [102]. However, Sharkey et al. indicated that SP600125 abolished the apoptosis of multiple myeloma cells [103]. These two studies clearly showed the controversial role of JNK in multiple myeloma through different mechanisms affecting cell apoptosis and survival. In the same way, the JNK pathway plays dual functions in prostate cancer [99]. JNK has been shown to contribute to the apoptosis of prostate cancer through endoplasmic reticulum stress, the death receptor-dependent apoptotic pathway, and the mitochondria pathway [99]. By contrast, several studies showed that the JNK pathway is involved in prostate cancer progression [99][104][105]. For example, Sung et al. found that Jazf1 promoted prostate cancer progression by activating JNK signaling in DU145 prostate cancer cells [104]. Similarly, Du et al. discovered that CC chemokine receptor 7 (CCR7) activated the Notch1 pathway, resulting in a significant increase of phosphorylated JNK, which further promoted the migration of prostate cancer cells [105]. In regard to oral cancer, the JNK pathway has been shown to play an oncogenic or tumor-suppressive role through action alone or synergistically with other MAPKs [106]. For instance, Noutomi et al. found that after activation, JNK further enhanced TRAIL-induced apoptosis of oral cancer cells [107]. Kim et al. discovered that JNK, together with ERK, were implicated in ROS-induced apoptosis of OSCC cells [108]. These studies showed that JNK has a tumor-inhibiting function in oral cancer. On the contrary, Gross et al. found that inhibition of JNK suppresses tumor growth in oral cancer by downregulating IL-8 signaling, VEGF activity, and EGFR activation [109], suggesting JNK’s pro-oncogenic role in oral cancer.
5.2. The Pro-Oncogenic Role of JNK in NSCLC and Glioblastoma
Emerging evidence suggests that the sustained activation of JNK contributes to the progression of NSCLC and glioblastoma [101]. Here, the researchers briefly discuss the role of JNK in NSCLC. Luo et al. found that LINC00958 promoted NSCLC cell proliferation by activating the JNK pathway [110]. Recently, Lin et al. reported that KIAA1429 promoted NSCLC tumorigenesis through the activation of the JNK pathway [111]. These studies showed a pro-oncogenic role of the JNK pathway in NSCLC (Figure 2B).
5.3. JNK Functions as a Tumor Suppressor in Intestinal Tumors, Papilloma, and Breast Cancer
The tumor-suppressive role of the JNK pathway in intestinal cancer, papilloma, and breast cancer is shown in Figure 2C. First, its role in breast cancer was supported by the findings of Cellurale and others that JNK1/2 ablation significantly increases tumor formation in breast cancer [112]. Subsequently, Shen et al. reported that cambogin induced the apoptosis of breast cancer cells by activating JNK signaling [113]. More recently, Itah et al. also found that JNK inhibition contributed to the progression of breast cancer [114]. Similarly, JNK also acts as a tumor suppressor in intestinal cancer and papilloma. For example, Tong et al. found that JNK1 knockout mice spontaneously developed intestinal cancer [115]. Recently, Kwak also reported that isolinderalactone exerted its anticancer effect on oxaliplatin-resistant colorectal cancer cells by inducing JNK-mediated apoptosis [116]. Choi et al. found that JNK1 can phosphorylate Myt1 to induce caspase-3-mediated apoptosis, thus preventing the formation of skin cancer, and that JNK1 knockout mice developed more UV-induced papilloma than JNK wild-type mice [117].
5.4. JNK Inactivation Suppresses Tumorigenesis in Ovarian Cancer, Skin Cancer, and Lymphoma
Emerging evidence showed that inhibition of JNK signaling prevented the oncogenesis of skin cancer, lymphoma, and ovarian cancer (Figure 2D) [101]. For instance, JNK2-KO mice exerted suppression of skin tumorigenesis [118], indicating the tumor-promoting role of JNK2 in skin cancer [100][118]. Additionally, Ding et al. found that targeting the JNK pathway suppressed Burkitt’s lymphoma cell proliferation [119]. Yang et al. found that JNK3 inactivation enhanced BH3 mimetic S1-induced apoptosis in cisplatin-resistant human ovarian cancer cells [120]. Similarly, JNK1 inhibition also suppressed ovarian cancer growth [121]. These results suggested the suppressive roles of different JNK isoforms in skin cancer, lymphoma, and ovarian cancer.
6. The JNK Pathway in Infectious Diseases
The JNK pathway has been shown to function in multiple infectious diseases, which are caused by pathogens, including viruses, bacteria, fungi, and parasites [122]. Thus, the researchers briefly discuss JNK’s role in these infectious diseases.
6.1. JNK in Viral Diseases
The JNK pathway can be activated by viruses, which, in turn, regulates many viral infectious diseases [123]. The majority of viruses use their encoding proteins to activate JNK signaling. For instance, the NS1 protein of the influenza A virus can activate JNK [124]. Similarly, the Tat protein of HIV-1 activates JNK signaling through a Nox-mediated mechanism [125]. The activated JNK, generally speaking, further supports viral infection and replication [122]. Previous studies have shown that JNK inactivation results in significantly reduced replication of viruses, such as dengue virus [126], influenza virus [127], and veterinary viruses [123]. Mechanistically, JNK promotes apoptosis of infected host cells, accelerating viral infection [122]. In addition, JNK is also important for viral replication via autophagy. In addition to the contribution of JNK to the replication of most viruses, JNKs also negatively regulate the replication of the oncolytic vaccinia virus [122].
6.2. JNK in Bacterial Infections
Previous studies have shown that JNK can be activated upon bacterial infection in multiple mammalian cell types [128][129]. Correspondingly, bacteria have evolved several mechanisms for activating JNK. For example, lipopolysaccharide (LPS), an outer membrane protein of Gram-negative bacteria, can activate the JNK signaling pathway [130]. Nguyen et al. showed that pneumolysin, a virulence factor of Streptococcus pneumoniae, can activate the JNK pathways to induce ATF3 expression [131]. Similarly, Escherichia coli (E. coli)-produced shiga toxin 1 is able to activate JNK, and subsequently cause the apoptosis of intestinal epithelial cells [132]. In addition, some bacteria themselves can regulate host JNK activity through posttranslational modification of JNK proteins [122]. For instance, AvrA protein, a salmonella effector protein, can promote acetylation of specific host MAPKKs, resulting in suppression of JNK signaling and ultimately hampering the host immune response against salmonella [133].
6.3. The JNK Pathway in Fungal and Parasitic Infections
The JNK pathway is activated upon fungal infection [122]. For example, JNK in human bronchial alveolar epithelial cells is activated by the virulence factor gliotoxin when infected with the mold Aspergillus fumigatus. Then the activated JNK mediates the apoptosis of human bronchial alveolar epithelial cells, ultimately causing invasive aspergillosis [134]. Additionally, the JNK pathway is also activated in host antifungal responses. An example is that Candida albicans-activated JNK1 signaling has a negative effect on antifungal innate immunity [135].
Similar to bacterial or fungal infections, JNK can be activated by parasitic infections. For example, JNK2 is activated by Toxoplasma gondii during the infection process [136]. Then, the activated JNK2 plays a dual role in host resistance and immunity [137].
7. The JNK Pathway in Other Diseases
JNK also functions in other diseases. Here, the researchers briefly discuss its role in other diseases that have not been mentioned above.
7.1. Hearing Loss
JNK is reported to be involved in deafness via promoting apoptosis [5]. Eshraghi et al. found that the JNK pathway is activated in hair cells under external stresses such as cochlear implantation trauma and noise. The activated JNK then induces hair cell apoptosis, ultimately causing deafness [138].
7.2. Ischemic/Reperfusion Injury
Ischemia/reperfusion injury often occurs in the brain, liver, heart, and kidneys. Although reperfusion is necessary for survival, it will result in tissue injury and activate inflammatory signaling, ultimately promoting JNK activation [6]. Taking myocardial infarction as an example, inflammatory signaling is stimulated in ischemic or necrotic myocardial cells, which ultimately leads to ventricular remodeling. This process involves ASK1-JNK1/2 signaling because ASK1 and JNK1/2 KO mice display reduced cardiac remodeling [6][139][140].
7.3. Cardiac Hypertrophy
Cardiac hypertrophy is defined as the thickening of the heart muscle under a variety of pathological stresses, such as mechanical stress and inflammation [6]. Although transient enlargement of the heart is a normal physiological phenomenon to adapt to these stimulations, the heart will suffer maladaptive hypertrophy under sustained pathological stresses, ultimately leading to heart failure [141]. The process of cardiac hypertrophy involves the activation of JNK signaling. For example, p-JNK is detected in patients with heart hypertrophy; therefore, JNK signaling is thought to promote myocyte growth, which marks pathologic hypertrophy [6].
7.4. Abdominal Aortic Aneurysms
Abdominal aortic aneurysms (AAAs) are one kind of arterial aneurysm. Chronic inflammation and vascular smooth muscle hypertrophy are considered the main reasons for AAA [6]. AAA formation involves JNK activation, as p-JNK is widely expressed in aneurysm tissue from patients with AAA [142]. Moreover, JNK expression in aneurysm tissue affects extracellular matrix metabolism, which further promotes the progression of AAA. Interestingly, pharmacological JNK inhibition alleviates AAA symptoms [142].
7.5. Renal Fibrosis
Renal fibrosis mainly refers to glomerular fibrosis and tubulointerstitial fibrosis, which ultimately develop into end-stage renal failure. These two processes involve activation of JNK signaling, as phosphorylated JNK is widely observed in glomerular cells and renal tissue from patients with acute and chronic renal injury [143]. Importantly, JNK inhibitors can ameliorate renal fibrosis. A key mechanism is that JNK can phosphorylate SMAD3, thus promoting the expression of pro-fibrotic genes, which further aggravate renal fibrosis [143].
7.6. Autosomal Dominant Polycystic Kidney Disease
Polycystic kidney disease (PKD) is a degenerative kidney disease wherein the renal tubules are filled with multiple fluid-filled cysts [144]. Autosomal dominant polycystic kidney disease (ADPKD) is one kind of PKD. ADPKD has been shown to be caused by mutations of the polycystin-1 (Pkd1) and Pkd2 genes. Smith et al. recently found that the JNK pathway is involved in ADPKD as inhibition of JNK activity reduces cyst growth [144].
References
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706.
- Hill, J.O. Understanding and addressing the epidemic of obesity: An energy balance perspective. Endocr. Rev. 2006, 27, 750–761.
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223.
- Pi-Sunyer, X. The medical risks of obesity. Postgrad. Med. 2009, 121, 21–33.
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348.
- Craige, S.M.; Chen, K.; Blanton, R.M.; Keaney, J.F., Jr.; Kant, S. JNK and cardiometabolic dysfunction. Biosci. Rep. 2019, 39, BSR20190267.
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336.
- Musi, N.; Goodyear, L.J. Insulin resistance and improvements in signal transduction. Endocrine 2006, 29, 73–80.
- Garg, R.; Kumariya, S.; Katekar, R.; Verma, S.; Goand, U.K.; Gayen, J.R. JNK signaling pathway in metabolic disorders: An emerging therapeutic target. Eur. J. Pharmacol. 2021, 901, 174079.
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371.
- Kant, S.; Barrett, T.; Vertii, A.; Noh, Y.H.; Jung, D.Y.; Kim, J.K.; Davis, R.J. Role of the mixed-lineage protein kinase pathway in the metabolic stress response to obesity. Cell Rep. 2013, 4, 681–688.
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184.
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783.
- Jaeschke, A.; Czech, M.P.; Davis, R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes. Dev. 2004, 18, 1976–1980.
- Menacho-Marquez, M.; Nogueiras, R.; Fabbiano, S.; Sauzeau, V.; Al-Massadi, O.; Dieguez, C.; Bustelo, X.R. Chronic sympathoexcitation through loss of Vav3, a Rac1 activator, results in divergent effects on metabolic syndrome and obesity depending on diet. Cell Metab. 2013, 18, 199–211.
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184.
- Tuncman, G.; Hirosumi, J.; Solinas, G.; Chang, L.; Karin, M.; Hotamisligil, G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 10741–10746.
- Waeber, G.; Delplanque, J.; Bonny, C.; Mooser, V.; Steinmann, M.; Widmann, C.; Maillard, A.; Miklossy, J.; Dina, C.; Hani, E.H.; et al. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat. Genet. 2000, 24, 291–295.
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91.
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611.
- Tanti, J.F.; Gremeaux, T.; van Obberghen, E.; Le Marchand-Brustel, Y. Serine/threonine phosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling. J. Biol. Chem. 1994, 269, 6051–6057.
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054.
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537.
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.S.; Karin, M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459.
- Sharfi, H.; Eldar-Finkelman, H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E307–E315.
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222.
- Maik-Rachline, G.; Wortzel, I.; Seger, R. Alternative Splicing of MAPKs in the Regulation of Signaling Specificity. Cells 2021, 10, 3466.
- Belgardt, B.F.; Mauer, J.; Wunderlich, F.T.; Ernst, M.B.; Pal, M.; Spohn, G.; Bronneke, H.S.; Brodesser, S.; Hampel, B.; Schauss, A.C.; et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 6028–6033.
- Sabio, G.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Ko, H.J.; Ong, H.; Morel, C.; Mora, A.; Reilly, J.; Kim, J.K.; et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes. Dev. 2010, 24, 256–264.
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Davis, R.J. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes. Dev. 2013, 27, 2345–2355.
- Vernia, S.; Morel, C.; Madara, J.C.; Cavanagh-Kyros, J.; Barrett, T.; Chase, K.; Kennedy, N.J.; Jung, D.Y.; Kim, J.K.; Aronin, N.; et al. Excitatory transmission onto AgRP neurons is regulated by cJun NH2-terminal kinase 3 in response to metabolic stress. Elife 2016, 5, e10031.
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525.
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Tournier, C.; Davis, R.J. Fibroblast Growth Factor 21 Mediates Glycemic Regulation by Hepatic JNK. Cell Rep. 2016, 14, 2273–2280.
- Lanuza-Masdeu, J.; Arevalo, M.I.; Vila, C.; Barbera, A.; Gomis, R.; Caelles, C. In vivo JNK activation in pancreatic beta-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes 2013, 62, 2308–2317.
- Hui, D.Y. A no-no for NonO and JNK in extracellular matrix homeostasis and vascular stability. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1677–1678.
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science 2004, 306, 1558–1561.
- Amini, N.; Boyle, J.J.; Moers, B.; Warboys, C.M.; Malik, T.H.; Zakkar, M.; Francis, S.E.; Mason, J.C.; Haskard, D.O.; Evans, P.C. Requirement of JNK1 for endothelial cell injury in atherogenesis. Atherosclerosis 2014, 235, 613–618.
- Min, R.W.M.; Aung, F.W.M.; Liu, B.; Arya, A.; Win, S. Mechanism and Therapeutic Targets of c-Jun-N-Terminal Kinases Activation in Nonalcoholic Fatty Liver Disease. Biomedicines 2022, 10, 2035.
- Czaja, M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol. Metab. 2010, 21, 707–713.
- Hirata, Y.; Inoue, A.; Suzuki, S.; Takahashi, M.; Matsui, R.; Kono, N.; Noguchi, T.; Matsuzawa, A. trans-Fatty acids facilitate DNA damage-induced apoptosis through the mitochondrial JNK-Sab-ROS positive feedback loop. Sci. Rep. 2020, 10, 2743.
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359.
- Jiang, Y.; Xu, J.; Huang, P.; Yang, L.; Liu, Y.; Li, Y.; Wang, J.; Song, H.; Zheng, P. Scoparone Improves Nonalcoholic Steatohepatitis Through Alleviating JNK/Sab Signaling Pathway-Mediated Mitochondrial Dysfunction. Front. Pharmacol. 2022, 13, 863756.
- Wei, Y.; Pagliassotti, M.J. Hepatospecific effects of fructose on c-jun NH2-terminal kinase: Implications for hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E926–E933.
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024.
- Wang, Q.; Zhou, H.; Bu, Q.; Wei, S.; Li, L.; Zhou, J.; Zhou, S.; Su, W.; Liu, M.; Liu, Z.; et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J. Hepatol. 2022, 77, 312–325.
- Guo, L.; Guo, Y.Y.; Li, B.Y.; Peng, W.Q.; Chang, X.X.; Gao, X.; Tang, Q.Q. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 2019, 294, 11805–11816.
- Win, S.; Than, T.A.; Kaplowitz, N. The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects. Int. J. Mol. Sci. 2018, 19, 3657.
- Schattenberg, J.; Czaja, M. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol. 2014, 3, 7–15.
- Wang, P.X.; Ji, Y.X.; Zhang, X.J.; Zhao, L.P.; Yan, Z.Z.; Zhang, P.; Shen, L.J.; Yang, X.; Fang, J.; Tian, S.; et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 2017, 23, 439–449.
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744.
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation Mediated by JNK in Myeloid Cells Promotes the Development of Hepatitis and Hepatocellular Carcinoma. Cell Rep. 2016, 15, 19–26.
- de Los Reyes Corrales, T.; Losada-Perez, M.; Casas-Tinto, S. JNK Pathway in CNS Pathologies. Int. J. Mol. Sci. 2021, 22, 3883.
- Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576.
- La Marca, J.E.; Richardson, H.E. Two-Faced: Roles of JNK Signalling During Tumourigenesis in the Drosophila Model. Front. Cell Dev. Biol. 2020, 8, 42.
- Musi, C.A.; Agro, G.; Santarella, F.; Iervasi, E.; Borsello, T. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells 2020, 9, 2190.
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tinto, S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019, 17, e3000545.
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281.
- Kitanaka, C.; Sato, A.; Okada, M. JNK Signaling in the Control of the Tumor-Initiating Capacity Associated with Cancer Stem Cells. Genes. Cancer 2013, 4, 388–396.
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516.
- Portela, M.; Mitchell, T.; Casas-Tinto, S. Cell-to-cell communication mediates glioblastoma progression in Drosophila. Biol. Open 2020, 9, bio053405.
- Jarabo, P.; de Pablo, C.; Herranz, H.; Martin, F.A.; Casas-Tinto, S. Insulin signaling mediates neurodegeneration in glioma. Life Sci. Alliance 2021, 4, e202000693.
- Hepp Rehfeldt, S.C.; Majolo, F.; Goettert, M.I.; Laufer, S. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases. Int. J. Mol. Sci. 2020, 21, 9677.
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005, 25, 1149–1158.
- Yoshida, H.; Hastie, C.J.; McLauchlan, H.; Cohen, P.; Goedert, M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 2004, 90, 352–358.
- Bozyczko-Coyne, D.; Saporito, M.S.; Hudkins, R.L. Targeting the JNK pathway for therapeutic benefit in CNS disease. Curr. Drug Targets CNS Neurol. Disord. 2002, 1, 31–49.
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560.
- Choi, W.S.; Abel, G.; Klintworth, H.; Flavell, R.A.; Xia, Z. JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J. Neuropathol. Exp. Neurol. 2010, 69, 511–520.
- Bekker, M.; Abrahams, S.; Loos, B.; Bardien, S. Can the interplay between autophagy and apoptosis be targeted as a novel therapy for Parkinson’s disease? Neurobiol. Aging 2021, 100, 91–105.
- Sieradzan, K.A.; Mann, D.M. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 1–21.
- Morfini, G.A.; You, Y.M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Bjorkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871.
- Perrin, V.; Dufour, N.; Raoul, C.; Hassig, R.; Brouillet, E.; Aebischer, P.; Luthi-Carter, R.; Deglon, N. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 2009, 215, 191–200.
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincon, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870.
- Zhang, P.; Miller, B.S.; Rosenzweig, S.A.; Bhat, N.R. Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J. Neurosci. Res. 1996, 46, 114–121.
- Bagnoud, M.; Briner, M.; Remlinger, J.; Meli, I.; Schuetz, S.; Pistor, M.; Salmen, A.; Chan, A.; Hoepner, R. c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis. Cells 2020, 9, 2154.
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2010, 2, 504–515.
- Singh, A.; Upadhayay, S.; Mehan, S. Understanding Abnormal c-JNK/p38MAPK Signaling Overactivation Involved in the Progression of Multiple Sclerosis: Possible Therapeutic Targets and Impact on Neurodegenerative Diseases. Neurotox. Res. 2021, 39, 1630–1650.
- Lindwall, C.; Kanje, M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell Neurosci. 2005, 29, 269–282.
- Pathak, A.; Clark, S.; Bronfman, F.C.; Deppmann, C.D.; Carter, B.D. Long-distance regressive signaling in neural development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2021, 10, e382.
- Macdonald, J.M.; Doherty, J.; Hackett, R.; Freeman, M.R. The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ. 2013, 20, 1140–1148.
- Huntwork-Rodriguez, S.; Wang, B.; Watkins, T.; Ghosh, A.S.; Pozniak, C.D.; Bustos, D.; Newton, K.; Kirkpatrick, D.S.; Lewcock, J.W. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 2013, 202, 747–763.
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 2015, 77, 13–25.
- Hirai, S.; Banba, Y.; Satake, T.; Ohno, S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J. Neurosci. 2011, 31, 6468–6480.
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931.
- Apara, A.; Galvao, J.; Wang, Y.; Blackmore, M.; Trillo, A.; Iwao, K.; Brown, D.P., Jr.; Fernandes, K.A.; Huang, A.; Nguyen, T.; et al. KLF9 and JNK3 Interact to Suppress Axon Regeneration in the Adult CNS. J. Neurosci. 2017, 37, 9632–9644.
- Lai, B.; Wu, C.H.; Lai, J.H. Activation of c-Jun N-Terminal Kinase, a Potential Therapeutic Target in Autoimmune Arthritis. Cells 2020, 9, 2466.
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001.
- Fukushima, A.; Boyle, D.L.; Corr, M.; Firestein, G.S. Kinetic analysis of synovial signalling and gene expression in animal models of arthritis. Ann. Rheum. Dis. 2010, 69, 918–923.
- Han, Z.; Boyle, D.L.; Chang, L.; Bennett, B.; Karin, M.; Yang, L.; Manning, A.M.; Firestein, G.S. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest. 2001, 108, 73–81.
- Schett, G.; Tohidast-Akrad, M.; Smolen, J.S.; Schmid, B.J.; Steiner, C.W.; Bitzan, P.; Zenz, P.; Redlich, K.; Xu, Q.; Steiner, G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 2501–2512.
- Lories, R.J.; Derese, I.; Luyten, F.P.; de Vlam, K. Activation of nuclear factor kappa B and mitogen activated protein kinases in psoriatic arthritis before and after etanercept treatment. Clin. Exp. Rheumatol. 2008, 26, 96–102.
- Li, X.; Wang, J.; Zhan, Z.; Li, S.; Zheng, Z.; Wang, T.; Zhang, K.; Pan, H.; Li, Z.; Zhang, N.; et al. Inflammation Intensity-Dependent Expression of Osteoinductive Wnt Proteins Is Critical for Ectopic New Bone Formation in Ankylosing Spondylitis. Arthritis Rheumatol. 2018, 70, 1056–1070.
- Ge, H.X.; Zou, F.M.; Li, Y.; Liu, A.M.; Tu, M. JNK pathway in osteoarthritis: Pathological and therapeutic aspects. J. Recept. Signal Transduct. Res. 2017, 37, 431–436.
- Im, H.J.; Muddasani, P.; Natarajan, V.; Schmid, T.M.; Block, J.A.; Davis, F.; van Wijnen, A.J.; Loeser, R.F. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cdelta pathways in human adult articular chondrocytes. J. Biol. Chem. 2007, 282, 11110–11121.
- Yang, P.; Tan, J.; Yuan, Z.; Meng, G.; Bi, L.; Liu, J. Expression profile of cytokines and chemokines in osteoarthritis patients: Proinflammatory roles for CXCL8 and CXCL11 to chondrocytes. Int. Immunopharmacol. 2016, 40, 16–23.
- Roy, P.K.; Rashid, F.; Bragg, J.; Ibdah, J.A. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 200–202.
- Mitsuyama, K.; Suzuki, A.; Tomiyasu, N.; Tsuruta, O.; Kitazaki, S.; Takeda, T.; Satoh, Y.; Bennett, B.L.; Toyonaga, A.; Sata, M. Pro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel disease. Int. J. Mol. Med. 2006, 17, 449–455.
- Chromik, A.M.; Muller, A.M.; Korner, J.; Belyaev, O.; Holland-Letz, T.; Schmitz, F.; Herdegen, T.; Uhl, W.; Mittelkotter, U. Genetic deletion of JNK1 and JNK2 aggravates the DSS-induced colitis in mice. J. Invest. Surg. 2007, 20, 23–33.
- Davies, C.; Tournier, C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 2012, 40, 85–89.
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679.
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857.
- Zhi, Y.; Zhou, X.; Yu, J.; Yuan, L.; Zhang, H.; Ng, D.C.H.; Xu, Z.; Xu, D. Pathophysiological Significance of WDR62 and JNK Signaling in Human Diseases. Front. Cell Dev. Biol. 2021, 9, 640753.
- Hideshima, T.; Hayashi, T.; Chauhan, D.; Akiyama, M.; Richardson, P.; Anderson, K. Biologic sequelae of c-Jun NH(2)-terminal kinase (JNK) activation in multiple myeloma cell lines. Oncogene 2003, 22, 8797–8801.
- Sharkey, J.; Khong, T.; Spencer, A. PKC412 demonstrates JNK-dependent activity against human multiple myeloma cells. Blood 2007, 109, 1712–1719.
- Sung, Y.; Park, S.; Park, S.J.; Jeong, J.; Choi, M.; Lee, J.; Kwon, W.; Jang, S.; Lee, M.H.; Kim, D.J.; et al. Jazf1 promotes prostate cancer progression by activating JNK/Slug. Oncotarget 2018, 9, 755–765.
- Du, R.; Tang, G.; Tang, Z.; Kuang, Y. Ectopic expression of CC chemokine receptor 7 promotes prostate cancer cells metastasis via Notch1 signaling. J. Cell Biochem. 2019, 120, 9639–9647.
- Gkouveris, I.; Nikitakis, N.G. Role of JNK signaling in oral cancer: A mini review. Tumour Biol. 2017, 39, 1010428317711659.
- Noutomi, T.; Itoh, M.; Toyota, H.; Takada, E.; Mizuguchi, J. Tumor necrosis factor-related apoptosis-inducing ligand induces apoptotic cell death through c-Jun NH2-terminal kinase activation in squamous cell carcinoma cells. Oncol. Rep. 2009, 22, 1169–1172.
- Kim, H.J.; Chakravarti, N.; Oridate, N.; Choe, C.; Claret, F.X.; Lotan, R. N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells. Oncogene 2006, 25, 2785–2794.
- Gross, N.D.; Boyle, J.O.; Du, B.; Kekatpure, V.D.; Lantowski, A.; Thaler, H.T.; Weksler, B.B.; Subbaramaiah, K.; Dannenberg, A.J. Inhibition of Jun NH2-terminal kinases suppresses the growth of experimental head and neck squamous cell carcinoma. Clin. Cancer Res. 2007, 13, 5910–5917.
- Luo, Z.; Han, Z.; Shou, F.; Li, Y.; Chen, Y. LINC00958 Accelerates Cell Proliferation and Migration in Non-Small Cell Lung Cancer Through JNK/c-JUN Signaling. Hum. Gene Ther. Methods 2019, 30, 226–234.
- Lin, X.; Ye, R.; Li, Z.; Zhang, B.; Huang, Y.; Du, J.; Wang, B.; Meng, H.; Xian, H.; Yang, X.; et al. KIAA1429 promotes tumorigenesis and gefitinib resistance in lung adenocarcinoma by activating the JNK/ MAPK pathway in an m(6)A-dependent manner. Drug Resist. Updat. 2023, 66, 100908.
- Cellurale, C.; Girnius, N.; Jiang, F.; Cavanagh-Kyros, J.; Lu, S.; Garlick, D.S.; Mercurio, A.M.; Davis, R.J. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2012, 72, 472–481.
- Shen, K.; Xie, J.; Wang, H.; Zhang, H.; Yu, M.; Lu, F.; Tan, H.; Xu, H. Cambogin Induces Caspase-Independent Apoptosis through the ROS/JNK Pathway and Epigenetic Regulation in Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 1738–1749.
- Itah, Z.; Chaudhry, S.; Raju Ponny, S.; Aydemir, O.; Lee, A.; Cavanagh-Kyros, J.; Tournier, C.; Muller, W.J.; Davis, R.J. HER2-driven breast cancer suppression by the JNK signaling pathway. Proc. Natl. Acad. Sci. USA 2023, 120, e2218373120.
- Tong, C.; Yin, Z.; Song, Z.; Dockendorff, A.; Huang, C.; Mariadason, J.; Flavell, R.A.; Davis, R.J.; Augenlicht, L.H.; Yang, W. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am. J. Pathol. 2007, 171, 297–303.
- Kwak, A.W.; Park, J.W.; Lee, S.O.; Lee, J.Y.; Seo, J.H.; Yoon, G.; Lee, M.H.; Choi, J.S.; Shim, J.H. Isolinderalactone sensitizes oxaliplatin-resistance colorectal cancer cells through JNK/p38 MAPK signaling pathways. Phytomedicine 2022, 105, 154383.
- Choi, H.S.; Bode, A.M.; Shim, J.H.; Lee, S.Y.; Dong, Z. c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer. Mol. Cell Biol. 2009, 29, 2168–2180.
- Chen, N.; Nomura, M.; She, Q.B.; Ma, W.Y.; Bode, A.M.; Wang, L.; Flavell, R.A.; Dong, Z. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res. 2001, 61, 3908–3912.
- Ding, X.; Wang, X.; Zhu, X.; Zhang, J.; Zhu, Y.; Shao, X.; Zhou, X. JNK/AP1 Pathway Regulates MYC Expression and BCR Signaling through Ig Enhancers in Burkitt Lymphoma Cells. J. Cancer 2020, 11, 610–618.
- Yang, X.; Xiang, X.; Xia, M.; Su, J.; Wu, Y.; Shen, L.; Xu, Y.; Sun, L. Inhibition of JNK3 promotes apoptosis induced by BH3 mimetic S1 in chemoresistant human ovarian cancer cells. Anat Rec (Hoboken) 2015, 298, 386–395.
- Vivas-Mejia, P.; Benito, J.M.; Fernandez, A.; Han, H.D.; Mangala, L.; Rodriguez-Aguayo, C.; Chavez-Reyes, A.; Lin, Y.G.; Carey, M.S.; Nick, A.M.; et al. c-Jun-NH2-kinase-1 inhibition leads to antitumor activity in ovarian cancer. Clin. Cancer Res. 2010, 16, 184–194.
- Chen, J.; Ye, C.; Wan, C.; Li, G.; Peng, L.; Peng, Y.; Fang, R. The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 9640.
- Wei, L.; Zhu, S.; Ruan, G.; Hou, L.; Wang, J.; Wang, B.; Liu, J. Infectious bursal disease virus-induced activation of JNK signaling pathway is required for virus replication and correlates with virus-induced apoptosis. Virology 2011, 420, 156–163.
- Nacken, W.; Wixler, V.; Ehrhardt, C.; Ludwig, S. Influenza A virus NS1 protein-induced JNK activation and apoptosis are not functionally linked. Cell Microbiol. 2017, 19, e12721.
- Wu, R.F.; Ma, Z.; Myers, D.P.; Terada, L.S. HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. J. Biol. Chem. 2007, 282, 37412–37419.
- Ceballos-Olvera, I.; Chavez-Salinas, S.; Medina, F.; Ludert, J.E.; del Angel, R.M. JNK phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 2010, 396, 30–36.
- Zhang, J.; Ruan, T.; Sheng, T.; Wang, J.; Sun, J.; Wang, J.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. Role of c-Jun terminal kinase (JNK) activation in influenza A virus-induced autophagy and replication. Virology 2019, 526, 1–12.
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252.
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40.
- Fu, J.; Peng, H. Lipopolysaccharides attenuates growth of HS cells through the JNK pathway. Cytotechnology 2016, 68, 2389–2394.
- Nguyen, C.T.; Kim, E.H.; Luong, T.T.; Pyo, S.; Rhee, D.K. TLR4 mediates pneumolysin-induced ATF3 expression through the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7 cells. Mol. Cells 2015, 38, 58–64.
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504.
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008, 3, 233–244.
- Geissler, A.; Haun, F.; Frank, D.O.; Wieland, K.; Simon, M.M.; Idzko, M.; Davis, R.J.; Maurer, U.; Borner, C. Apoptosis induced by the fungal pathogen gliotoxin requires a triple phosphorylation of Bim by JNK. Cell Death Differ. 2013, 20, 1317–1329.
- Zhao, X.; Guo, Y.; Jiang, C.; Chang, Q.; Zhang, S.; Luo, T.; Zhang, B.; Jia, X.; Hung, M.C.; Dong, C.; et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat. Med. 2017, 23, 337–346.
- Valere, A.; Garnotel, R.; Villena, I.; Guenounou, M.; Pinon, J.M.; Aubert, D. Activation of the cellular mitogen-activated protein kinase pathways ERK, P38 and JNK during Toxoplasma gondii invasion. Parasite 2003, 10, 59–64.
- Sukhumavasi, W.; Warren, A.L.; Del Rio, L.; Denkers, E.Y. Absence of mitogen-activated protein kinase family member c-Jun N-terminal kinase-2 enhances resistance to Toxoplasma gondii. Exp. Parasitol. 2010, 126, 415–420.
- Eshraghi, A.A.; Van de Water, T.R. Cochlear implantation trauma and noise-induced hearing loss: Apoptosis and therapeutic strategies. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 473–481.
- Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Kashiwase, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Watanabe, T.; Asahi, M.; Taniike, M.; et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 15883–15888.
- Izumiya, Y.; Kim, S.; Izumi, Y.; Yoshida, K.; Yoshiyama, M.; Matsuzawa, A.; Ichijo, H.; Iwao, H. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ. Res. 2003, 93, 874–883.
- Dickhout, J.G.; Carlisle, R.E.; Austin, R.C. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: Endoplasmic reticulum stress as a mediator of pathogenesis. Circ. Res. 2011, 108, 629–642.
- Yoshimura, K.; Aoki, H.; Ikeda, Y.; Fujii, K.; Akiyama, N.; Furutani, A.; Hoshii, Y.; Tanaka, N.; Ricci, R.; Ishihara, T.; et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat. Med. 2005, 11, 1330–1338.
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK Signaling Pathway in Renal Fibrosis. Front. Physiol. 2017, 8, 829.
- Smith, A.O.; Jonassen, J.A.; Preval, K.M.; Davis, R.J.; Pazour, G.J. c-JUN n-Terminal Kinase (JNK) Signaling in Autosomal Dominant Polycystic Kidney Disease. J. Cell Signal 2022, 3, 62–78.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
874
Revisions:
2 times
(View History)
Update Date:
11 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No