Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vishwanath Venketaraman | -- | 3090 | 2024-03-07 06:16:50 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dawi, J.; Mohan, A.S.; Misakyan, Y.; Affa, S.; Gonzalez, E.; Hajjar, K.; Nikoghosyan, D.; Fardeheb, S.; Tuohino, C.; Venketaraman, V. Role of Oxidative Stress in Tuberculous Meningitis. Encyclopedia. Available online: https://encyclopedia.pub/entry/55949 (accessed on 25 July 2026).
Dawi J, Mohan AS, Misakyan Y, Affa S, Gonzalez E, Hajjar K, et al. Role of Oxidative Stress in Tuberculous Meningitis. Encyclopedia. Available at: https://encyclopedia.pub/entry/55949. Accessed July 25, 2026.
Dawi, John, Aishvaryaa Shree Mohan, Yura Misakyan, Scarlet Affa, Edgar Gonzalez, Karim Hajjar, David Nikoghosyan, Sabrina Fardeheb, Christopher Tuohino, Vishwanath Venketaraman. "Role of Oxidative Stress in Tuberculous Meningitis" Encyclopedia, https://encyclopedia.pub/entry/55949 (accessed July 25, 2026).
Dawi, J., Mohan, A.S., Misakyan, Y., Affa, S., Gonzalez, E., Hajjar, K., Nikoghosyan, D., Fardeheb, S., Tuohino, C., & Venketaraman, V. (2024, March 07). Role of Oxidative Stress in Tuberculous Meningitis. In Encyclopedia. https://encyclopedia.pub/entry/55949
Dawi, John, et al. "Role of Oxidative Stress in Tuberculous Meningitis." Encyclopedia. Web. 07 March, 2024.
Copy Citation
Meningitis is an inflammatory condition affecting the meninges surrounding the brain and spinal cord. Meningitis can be triggered by various factors, including infectious agents like viruses and bacteria and non-infectious contributors such as cancer or head injuries. The impact of meningitis on the central nervous system involves disruptions in the blood–brain barrier, cellular infiltrations, and structural alterations.
tuberculous meningitis
non-tuberculous meningitis
reactive oxygen species
ferroptosis
1. Introduction
1.1. Molecular Bases of Meningitis Pathophysiology
Meningitis, irrespective of etiology, involves complex molecular interactions between the host and various infectious agents. The interplay of cytokines, chemokines, and immune cells orchestrates the inflammatory cascade. To begin with, the blood–brain barrier (BBB) relies on a complex interplay within the neurovascular unit, including brain endothelium, pericytes, astrocytes, and basal membranes. Brain endothelial cells maintain brain homeostasis through membrane transporters like glucose transporter 1 (GLUT1) and P-glycoprotein (Pgp), while tight junctions (TJs) prevent paracellular diffusion, ensuring cellular polarity. The glycocalyx on the luminal side and the basement membrane on the basolateral side contribute to barrier integrity. Brain microvascular endothelial cells exhibit a tight barrier, but post-capillary venules and veins have “leaky” junctions, potentially serving as a passage for pathogens into the cerebrospinal fluid [1][2][3][4].
Secondly, the blood–cerebrospinal fluid barrier (BCSFB) at the choroid plexus is formed by choroidal epithelial cells facing blood and cerebrospinal fluid (CSF). These cells control fenestration, allowing exchange between blood components and stromal tissue. ATP-binding cassette transporters (ABC), solute carrier transporters, and tight-junction complexes regulate barrier permeability. Choroidal epithelial cells produce CSF, and their strategic localization allows the integration of signals from blood and the brain. The choroid plexus contributes to neuroimmune surveillance and immune cell trafficking into the central nervous system [2][5].
In addition, the meninges covering the central nervous system (CNS) consist of the dura mater, arachnoid mater, and pia mater. The arachnoid mater primarily forms the BCSFB function, connecting leptomeningeal cells through tight junctions. Leptomeninges play a crucial role in bacterial meningitis, and an additional BCSFB function is proposed for pial vessels. Like the choroid plexus, the arachnoid regulates CNS immunity through major histocompatibility complex (MHC) class II-expressing myeloid cells. Macrophages and dendritic cells on the dura-facing side and resident macrophages on the CSF-facing side contribute to immune surveillance and potential antigen presentation to T cells [2][6][7][8] (Figure 1).
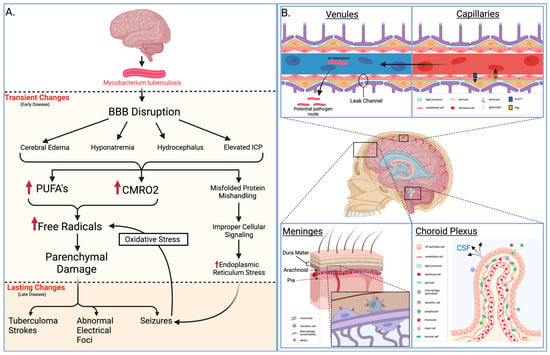
Figure 1. Pathology of meningitis. (A) Early and late pathological changes that result from M. tuberculosis meningitis. (B) Neurological and vascular structures displaying one infectious route, key immunological defenses, and relevant histology. BBB = blood–brain barrier, PUFA = polyunsaturated fatty acid, CMRO2 = cerebral metabolic rate of oxygen, GLUT1 = glucose transporter 1, Pgp = P-glycoprotein, MHC2 = major histocompatibility complex class II, CP = choroid plexus, CSF = cerebrospinal fluid.
1.2. Pathology of Meningitis
Understanding the pathological changes induced by meningitis is crucial for unraveling its impact on the central nervous system. For example, pathogens like tuberculous meningitis (TBM) cause such a disruption, and seizures may occur due to various factors influenced by diverse pathological changes. Some of these changes are transient and can be resolved with appropriate measures, while others may persist, necessitating prolonged treatment with anti-epileptic drugs. Transient causes, often encountered in the early clinical phase, include meningeal irritation, cerebral edema, hyponatremia, hydrocephalus, and elevated intracranial pressure. In the later stages of TBM, multiple intracranial tuberculoma strokes, the development of abnormal electric foci, and other unidentified causes may contribute to seizures that pose challenges in terms of management. Seizures have the potential to induce oxidative stress in the brain parenchyma.
In various central nervous system pathologies, excessive production of free radicals or diminished antioxidant activity can precipitate seizures, heightening the likelihood of recurrence. Conversely, seizures can also arise as a consequence of oxidative stress. Improper handling of misfolded proteins by the endoplasmic reticulum, leading to their accumulation, may result in cellular dysfunction and cell death by activating diverse signal transduction pathways. Impaired protein signaling can disrupt the translocation and transcription of proteins via pathways like the inositol pathway, which involves 1-α protein kinase-like ER kinase and activating transcriptional factor 6 (ATF-6). Endoplasmic reticulum stress (ER stress) is mitigated by unfolded protein response (UPR) through the involvement of the transcriptional factor X-box binding protein (XBP).
Seizures observed in TBM may be linked to heightened oxidative stress and endoplasmic reticulum (ER) stress, potentially influencing the severity of meningitis and impacting patient outcomes. Understanding the substantial impact of these changes in TBM could unveil potential therapeutic targets. Oxidative stress and ER stress concerning seizures associated with TBM have yet to be thoroughly examined [9].
1.3. US Statistics of Meningitis
In the United States, the annual toll of meningitis unfolds through more than 72,000 hospitalizations, incurring a substantial financial burden exceeding 1.2 billion USD [10]. Diverse strains of infectious meningitis cast shadows of heightened mortality and enduring complexities, encompassing neurological deficits and cognitive impairments [11][12]. Bacterial meningitis, a formidable adversary, wields mortality rates ranging from 10% to 20% in well-resourced settings and up to 50% in regions with fewer resources, where it assumes the somber rank of the fourth-leading cause of disability [11][12][13][14].
Parallel to this, fungal meningitis, notably cryptococcal meningitis, manifests with a poignant in-hospital mortality ranging between 30% and 50% [15]. On the global stage, Cryptococcus-induced meningitis bears the weight of 15% of AIDS-related deaths [16]. The specter of mortality also shrouds other rare forms of fungal meningitis. As Antinori et al. reported, Aspergillus meningitis carries a case fatality rate of 63.5% for those with intact immune defenses and a staggering 83% for immunocompromised patients [17]. Meningitis kindled by Coccidioides signals an unremitting journey toward mortality, hitting 90% at the first year’s end and an unrelenting 100% by the end of the second year without intervention [18]. Finally, central nervous system infections orchestrated by Histoplasma have a 39% case fatality rate [19].
1.4. Global Statistics of Meningitis
With the global goal of reducing meningitis by 2030, it has become a prevalent issue to defeat. According to the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD), in 2019, there were an estimated 236,000 deaths due to meningitis, with the largest burden in children under the age of five, with 112,000 deaths [20]. Regions with the largest rate of meningitis include western sub-Saharan Africa, followed by eastern sub-Saharan Africa, central and south sub-Saharan Africa, and Oceania [21].
Although globally, there is a downward trend in meningitis incidence, with 7.5 per 100,000 in 1990 to 3.3 per 100,000 in 2019, when looking at individual countries, the rates seem to be increasing, as in sub-Saharan Africa and Oceania [20]. As pediatric populations are the most susceptible, there is a higher prevalence among this age group and thus a higher mortality of 20–30% [21].
2. Tuberculous Meningitis and Non-Tuberculous Meningitis
Numerous pathogens can cause meningitis. Differentiating between the etiologies of meningitis can be challenging, but TBM has certain clinical features that can assist in diagnosis. These include positive CSF cultures for Mycobacterium tuberculosis (Mtb) and smears for acid-fast bacilli (AFB), basal enhancement on CT scan, and response to antituberculous treatment [22]. Non-tuberculous meningitis (NTM) primarily has bacterial, viral, or fungal etiology.
The disease process of bacterial meningitis involves several stages: mucosal colonization, systemic invasion, survival within the bloodstream and meninges, and neuronal damage due to increased intracranial pressure and altered cerebral blood flow [23]. The process begins with the colonization of the nasopharynx by the pathogen. This is followed by systemic invasion, leading to bacteremia. The bacterial encapsulation helps the pathogen resist phagocytosis and complement-mediated bactericidal activity, thus contributing to bacteremia. CNS invasion occurs after that, although the precise site of bacterial traversal into the CNS remains unknown. Bacterial replication and lysis in the subarachnoid space release virulence factors, inciting an inflammatory response in the CNS. This inflammation, marked by cytokine and chemokine release, leads to leukocyte influx, exacerbating brain damage.
In contrast, anti-inflammatory proteins like interleukin-10 (IL-10) and transforming growth factor beta 1 (TGF-β1) help regulate this inflammatory activity [24]. Clinically, bacterial meningitis patients often present with fever, headache, and signs of cerebral dysfunction. The classic triad of fever, neck stiffness, and altered mental status is not always present. The disease can progress to seizures, focal neurological deficits, and increased intracranial pressure, with pneumococcal and meningococcal meningitis occasionally presenting with rapid sepsis.
Viruses responsible for meningitis are typically transmitted through inhalation, as seen with mumps, or ingestion, as with non-polio enteroviruses, and initially establish a primary infection in the body’s oropharyngeal or gastrointestinal lymphoid tissues. Once in the body, these pathogens can access the central nervous system (CNS) by several routes: they may infect cerebral vascular endothelial cells, cross the BBB by infecting hematopoietic cells, or travel via peripheral sensory or motor neurons. Upon reaching the CNS and establishing infection, the viral presence triggers the release of chemoattractants within the meninges, prompting an innate immune response marked by the infiltration of neutrophils, monocytes, and antiviral cluster of differentiation 8 (CD8) lymphocytes. This immune response to the viral invasion is a crucial feature of the pathogenesis of viral meningitis.
Fungal meningitis, although less common, is a significant concern, especially in immunocompromised individuals. It usually presents as a subacute or chronic process and can be as lethal as bacterial meningitis if left untreated. Most pathogenic fungi are inhaled, leading to a primary pulmonary infection, usually self-limited. Hematogenous dissemination may follow, with subsequent involvement of the CNS. The subarachnoid space and its contents are usually immunologically protected, with functional and anatomic barriers to invasion. In immunocompromised individuals, however, the dysfunctional protective structures allow penetration of fungal pathogens into the CNS [10]. Cryptococcal meningitis (CrM), predominantly caused by Cryptococcus neoformans or Cryptococcus gattii, is the leading cause of adult fungal meningitis and is increasingly a global health concern. HIV-infected individuals are at the greatest risk, with CrM implicated in up to 79% of cases within this group. CrM is responsible for a significant proportion of AIDS-related deaths—between 15% and 17%—even in regions with adequate health-care resources [25].
TBM’s global incidence and mortality are not well documented due to diagnostic difficulties and inconsistent reporting practices [26]. It is estimated that in 2019, approximately 164,000 adults worldwide developed TBM, with a significant 23% co-infected with HIV. Predominantly, cases were male (60%) and in the 25–34 age bracket (20%), with the majority of cases (70%) occurring in Southeast Asia and Africa. The study estimated that in the same year, 78,200 adults succumbed to TBM, which is about 48% of those afflicted [27]. The study, however, is not without limitations. Data variation due to different study designs, inclusion criteria, and TBM definitions could affect the estimates’ accuracy. The extrapolation of data to all countries may not account for local factors such as TB prevalence, population demographics, genetics, comorbidities, or the efficacy of health systems. The study also presumed that the proportion of undiagnosed TBM was equivalent to that of diagnosed cases, a supposition not backed by specific data, leading to potential inaccuracies. TBM stands as the deadliest manifestation of tuberculosis, and it is clear that there needs to be an improvement in TBM monitoring and treatment globally.
NTM and TBM present distinct pathological features and clinical progressions. NTM symptoms can vary, but include fever, headache, stiff neck, nausea, vomiting, photophobia, and altered mental status [28]. NTM is most often caused by bacteria like Streptococcus pneumoniae and viruses such as enteroviruses, which lead to an acute inflammatory response in the meninges. The inflammation is typically characterized by rapid onset and, in bacterial forms, is associated with purulent CSF.
Conversely, TBM follows a more protracted course, often beginning with a subacute phase that may last for weeks. TBM symptoms include fever, headache, stiff neck, confusion, altered mental status, and neurological deficits [29]. The concept of a “Rich” focus, described by Rich and McCordock, seems to also play a part in TBM pathogenesis. They suggest that initial TB infection may lead to silent tuberculous lesions past the BBB. Afterward, activation of previously dormant tuberculous lesions releases the bacteria into the subarachnoid space, inciting a granulomatous infection of the meninges and consequential inflammatory response [30]. This inflammation can account for various clinical manifestations of TBM.
3. Oxidative Stress in Tuberculosis
TB and NTM pose significant global health challenges due to their intricate pathogenesis and the complex interplay between infecting agents and the host’s immune response. Recent scientific endeavors have shed light on the crucial role of oxidative stress and reactive species in these infectious diseases. Oxidative stress, mediated by reactive oxygen species (ROS), ferroptosis, and reactive nitrogen species (RNS), has emerged as a key contributor to the progression, severity, and modulation of the immune response in TB and NTM.
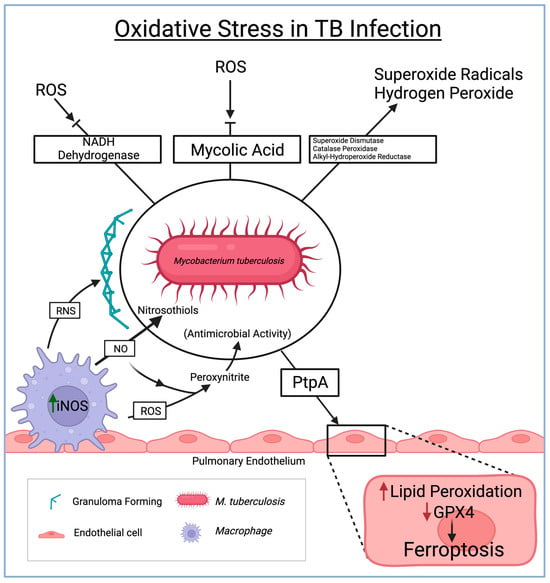
Reactive oxygen species (ROS) play a critical role in bolstering the host’s defense against TBM. However, the TB-causing Mycobacterium tuberculosis (Mtb) has developed mechanisms to counteract and evade the effects of ROS. Particularly within the CNS, Mtb produces antioxidant enzymes like superoxide dismutase, catalase-peroxidase, and alkyl hydroperoxide reductase. These enzymes effectively neutralize ROS, including superoxide radicals and hydrogen peroxide, prevalent within the CNS environment. By breaking down ROS, the bacteria can thrive amidst the oxidative stress within the CNS [31].
Ferroptosis, a newly explored phenomenon, is gaining attention in the realm of tuberculous meningitis. Ferroptosis, an iron-dependent form of regulated cell death characterized by lipid peroxidation and the accumulation of toxic lipid reactive intermediates, is being increasingly studied within the context of TBM. Mtb has evolved mechanisms to acquire iron from the host within the CNS, leading to an accumulation of excess iron, thereby creating a conducive environment for ferroptosis [32]. Among the seven identified glutathione peroxidase (GPX) enzymes, GPX4 is most closely associated with the ferroptosis pathways [33].
Moreover, the study revealed that Mtb lacking protein tyrosine phosphatase A (PtpA), which serves as a pro-ferroptosis factor, exhibited a reduced capability to lower GPX4 expression compared to the wild-type Mtb. This finding further supports that PtpA is a crucial effector in Mtb, inhibiting GPX4 expression and encouraging ferroptosis in host cells [34][35]. However, the detailed interplay between ferroptosis and TBM remains a subject of ongoing research, requiring further exploration for a comprehensive understanding.
Reactive nitrogen species (RNS) also significantly contribute to the immune response against Mtb within the CNS during TBM. Nitric oxide, a key RNS, reacts with superoxide anions to form highly reactive nitrogen intermediates such as peroxynitrite [29]. These intermediates function as potent antimicrobial agents, inflicting damage upon Mtb. Furthermore, nitric oxide disrupts Mtb survival mechanisms by reacting with thiol groups in proteins, leading to the formation of nitrosothiols. RNS also plays a pivotal role in granuloma formation (the organized aggregation of immune cells containing Mtb within the CNS), which is crucial in preventing the spread of TBM [36] (Figure 2).
Figure 2. The role of oxidative stress in the immune system and consequent defenses by M. tuberculosis in driving pathological routes to endothelial damage and granuloma formation. ROS = reactive oxygen species, TB = tuberculosis, NADH = nicotinamide adenine dinucleotide + hydrogen, RNS = reactive nitrogen species, PtpA = protein tyrosine phosphatase A, GPX4 = glutathione peroxidase 4.
4. TB Meningitis Treatment Options
In order to effectively treat TBM, it is imperative to examine three components for successful management: (i) rapidly diminishing the actively multiplying bacilli to reduce disease severity, mortality, and transmission, (ii) eliminating populations of lingering bacilli to ensure a lasting cure and prevent relapse, and (iii) averting the development of drug resistance throughout treatment [37]. Initiating treatment for TBM is crucial at the earliest signs of clinical suspicion, even before obtaining microbiological confirmation through molecular tests, mycobacterial culture, and acid-fast bacilli smear microscopy [37][38].
In all forms of tuberculosis, INH serves as a crucial chemotherapeutic agent in the treatment of TBM, as it quickly reaches the CNS, rapidly reduces mortality, and demonstrates robust bactericidal activity [38][39][40]. In contrast, RMP does not freely penetrate the BBB as effectively, with 10–20% of its concentration in the CSF, and only becomes activated when unbound to protein [39]. However, patients with RMP-resistant strains experience an increased mortality to TBM, confirming its role in the treatment protocol [38]. PZE has effective CNS penetration, with similar concentrations in the CSF and serum, and reduces the duration of treatment for drug-susceptible TB [38][39]. Although its bactericidal activity is diminished in the first 2–4 days of treatment, it reaches potency similar to INH and RMP in days 4–14 [39]. The last of the RIPE regimens, SM or ETB, has the poorest CNS penetration and provokes significant adverse effects with long-term use, which leads researchers to investigate the use of a fourth drug [38][39][40]. However, with the significant rise in RPM-resistant (RR) and multidrug-resistant TB (MDR-TB), further research is implicated to successfully optimize patient outcomes.
Although antibiotic therapy overall reduces morbidity, its variable penetration in the brain and severe inflammatory response renders it ineffective at curing TBM [41]. In addition to the RIPE protocol, fluoroquinolones are incorporated to control TBM disruption and mitigate multidrug-resistant cases. The newer generation of fluoroquinolones, such as levofloxacin and moxifloxacin, exhibited highly potent activity against most Mtb strains, excellent CSF penetration, and favorable safety profiles [38].
The neurological aspects of TBM manifest as an overactive inflammatory response that extensively damages tissue and confines the brain to a fixed space [38][41]. Therefore, there has been considerable attention on supplementary host-directed immune interventions to boost protective immunity, limit neurological complications, or modulate tissue destruction [39][42].
5. Alternative Therapy for Oxidative Stress-Induced TB Meningitis
The management of TBM relies on eliminating Mtb and managing host inflammatory responses [43]. Alternative treatment of oxidative stress-induced TB meningitis heavily depends on eliminating inflammatory processes such as ferroptosis. As previously established, oxidative stress induces ferroptosis through the overload of reactive oxygen species (ROS), which leads to lipid peroxidation and glutathione (GSH) depletion; hence, ferroptosis plays a key role in oxidative stress-induced TB meningitis [44]. As the disease takes its course, ferroptosis simultaneously occurs.
Iron chelators are a prevalent and continuously studied therapy for ferroptosis. The FDA has approved three main iron chelators for clinical administration: deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX) [45]. Iron chelators such as DFO or DFP work by binding to the accumulated iron to prevent mitochondrial ROS accumulation [46]. They can also remove iron from lipid-peroxidizing enzymes, specifically lipoxygenases (LOXs), which are fundamental to ferroptosis mechanisms [46].
Nonetheless, DFO poses numerous clinical limitations and side effects, such as skin, ocular, and auditory reactions, with neurological and pulmonary disorders being observed at high doses [46]. DFP, on the other hand, is orally administered in tablet or solution form. The dosage for adults and pediatric patients is 75 mg/kg per day divided over three separate doses. The recommended dose is 100 mg/kg daily [46].
Moreover, another meta-analysis of 16 RCTs concluded that patients who received doses less than 30 mg/kg daily and for a period shorter than 12 weeks demonstrated significant decreases in MDA levels [47]. Other markers of oxidative stress, such as total oxidant status (TOS), TAC, glutathione peroxidase (GPX), superoxide dismutase (SOD), and prooxidant–antioxidant balance (PAB), were also impacted in an oxidative stress-reducing manner. However, the conditions under which this occurred contradicted previously outlined treatment protocols [47]. Given this ambiguity, RCTs with larger samples must be carried out to assess saffron’s effects on various oxidative stress markers, its optimal dosing, and its role in therapeutic endeavors for oxidative stress-induced TB meningitis.
References
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the Blood–Brain barriers and Blood–Brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314.
- Dias, M.C.; Mapunda, J.A.; Vladymyrov, M.; Engelhardt, B. Structure and junctional complexes of endothelial, epithelial and glial brain barriers. Int. J. Mol. Sci. 2019, 20, 5372.
- Butt, A.M.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62.
- Join-Lambert, O.; Morand, P.; Carbonnelle, É.; Coureuil, M.; Bille, E.; Bourdoulous, S.; Nassif, X. Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog. Neurobiol. 2010, 91, 130–139.
- Ghersi-Egea, J.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361.
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Møllgård, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385.
- Christodoulides, M.; Makepeace, B.L.; Partridge, K.; Kaur, D.; Fowler, M.; Weller, R.O.; Heckels, J.E. Interaction of Neisseria meningitidis with Human Meningeal Cells Induces the Secretion of a Distinct Group of Chemotactic, Proinflammatory, and Growth-Factor Cytokines. Infect. Immun. 2002, 70, 4035–4044.
- Rua, R.; McGavern, D.B. Advances in meningeal immunity. Trends Mol. Med. 2018, 24, 542–559.
- Misra, U.K.; Tripathi, A.; Kumar, M. Oxidative and endoplasmic reticulum stress in tuberculous meningitis related seizures. Epilepsy Res. 2019, 156, 106160.
- Charalambous, L.T.; Premji, A.; Tybout, C.; Hunt, A.; Cutshaw, D.; Elsamadicy, A.A.; Yang, S.; Xie, J.; Giamberardino, C.; Pagadala, P.; et al. Prevalence, healthcare resource utilization and overall burden of fungal meningitis in the United States. J. Med. Microbiol. 2018, 67, 215–227.
- Bhimraj, A. Acute community-acquired bacterial meningitis in adults: An evidence-based review. Clevel. Clin. J. Med. 2012, 79, 393–400.
- Mount, H.R.; Boyle, S.D. Aseptic and bacterial meningitis: Evaluation, treatment, and prevention. PubMed 2017, 96, 314–322.
- Amidu, N.; Antuamwine, B.B.; Addai-Mensah, O.; Abdul-Karim, A.; Azure, S.; Abubakari, B.B.; Abenyeri, J.; Opoku, A.S.; Nkukah, J.E.; Najibullah, A.S. Diagnosis of bacterial meningitis in Ghana: Polymerase chain reaction versus latex agglutination methods. PLoS ONE 2019, 14, e0210812.
- Houri, H.; Pormohammad, A.; Riahi, S.M.; Nasiri, M.J.; Fallah, F.; Dabiri, H.; Pouriran, R. Acute bacterial meningitis in Iran: Systematic review and meta-analysis. PLoS ONE 2017, 12, e0169617.
- Abassi, M.; Boulware, D.R.; Rhein, J. Cryptococcal meningitis: Diagnosis and Management Update. Curr. Trop. Med. Rep. 2015, 2, 90–99.
- Rajasingham, R.; Smith, R.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881.
- Antinori, S.; Corbellino, M.; Meroni, L.; Resta, F.; Sollima, S.; Tonolini, M.; Tortorano, A.M.; Milazzo, L.; Bello, L.; Furfaro, E.; et al. Aspergillus meningitis: A rare clinical manifestation of central nervous system aspergillosis. Case report and review of 92 cases. J. Infect. 2013, 66, 218–238.
- Stevens, D.A.; Zhang, Y.; Finkelman, M.; Pappagianis, D.; Clemons, K.V.; Martinez, M. Cerebrospinal Fluid (1,3)-Beta-d-Glucan Testing Is Useful in Diagnosis of Coccidioidal Meningitis. J. Clin. Microbiol. 2016, 54, 2707–2710.
- Wheat, L.J.; Batteiger, B.E.; Sathapatayavongs, B. Histoplasma capsulatum Infections of the Central Nervous System. Medicine 1990, 69, 244.
- Wunrow, H.Y.; Bender, R.G.; Vongpradith, A.; Sirota, S.B.; Swetschinski, L.R.; Novotney, A.; Gray, A.P.; Ikuta, K.S.; Sharara, F.; Wool, E.E.; et al. Global, regional, and national burden of meningitis and its aetiologies, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2023, 22, 685–711.
- Kwambana-Adams, B. Global burden of meningitis and implications for strategy. Lancet Neurol. 2023, 22, 646–648.
- Kumar, R.; Singh, S.; Kohli, N. A diagnostic rule for tuberculous meningitis. Arch. Dis. Child. 1999, 81, 221–224.
- Nudelman, Y.; Tunkel, A.R. Bacterial meningitis. Drugs 2009, 69, 2577–2596.
- Van Furth, A.M.; Seijmonsbergen, E.M.; Langermans, J.A.M.; Groeneveld, P.H.P.; De Bel, C.E.; Van Furth, R. High levels of interleukin 10 and tumor necrosis factor in cerebrospinal fluid during the onset of bacterial meningitis. Clin. Infect. Dis. 1995, 21, 220–222.
- Williamson, P.R.; Jarvis, J.N.; Panackal, A.A.; Fisher, M.C.; Molloy, S.F.; Loyse, A.; Harrison, T.S. Cryptococcal meningitis: Epidemiology, immunology, diagnosis and therapy. Nat. Rev. Neurol. 2016, 13, 13–24.
- Seddon, J.A.; Tugume, L.; Solomons, R.; Prasad, K.; Bahr, N.C. The current global situation for tuberculous meningitis: Epidemiology, diagnostics, treatment and outcomes. Wellcome Open Res. 2019, 4, 167.
- Dodd, P.J.; Osman, M.; Cresswell, F.V.; Stadelman, A.; Lân, N.H.; Thuong, N.T.T.; Muzyamba, M.; Glaser, L.; Dlamini, S.N.; Seddon, J.A. The global burden of tuberculous meningitis in adults: A modelling study. PLoS Glob. Public Health 2021, 1, e0000069.
- Jung, J.; Shin, I.; Choi, Y. A Rare Case of Nontuberculous Mycobacterial Abscess Mimicking Brain Tumor in an Immunocompetent Patient. Brain Tumor Res. Treat. 2023, 11, 219–222.
- Arshad, A.; Dayal, S.; Gadhe, R.; Mawley, A.; Shin, K.; Tellez, D.; Phan, P.; Venketaraman, V. Analysis of tuberculosis meningitis Pathogenesis, diagnosis, and treatment. J. Clin. Med. 2020, 9, 2962.
- Donald, P.R.; Schaaf, H.S.; Schoeman, J. Tuberculous meningitis and miliary tuberculosis: The Rich focus revisited. J. Infect. 2005, 50, 193–195.
- Shastri, M.D.; Shukla, S.D.; Chong, W.C.; Dua, K.; Peterson, G.; Patel, R.; Hansbro, P.M.; Eri, R.; O’Toole, R. Role of oxidative stress in the pathology and management of human tuberculosis. Oxidative Med. Cell. Longev. 2018, 2018, 7695364.
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J. Ferroptosis: A mixed blessing for infectious diseases. Front. Pharmacol. 2022, 13, 992734.
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In Apoptotic and Non-Apoptotic Cell Death; Nagata, S., Nakano, H., Eds.; Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2016; Volume 403.
- Liang, T.; Chen, J.; Xu, G.; Zhang, Z.; Xue, J.; Zeng, H.; Jiang, J.; Chen, T.; Qin, Z.; Li, H.; et al. Ferroptosis-related gene SOCS1, a marker for tuberculosis diagnosis and treatment, involves in macrophage polarization and facilitates bone destruction in tuberculosis. Tuberculosis 2022, 132, 102140.
- Gan, B. Ferroptosis hijacking by Mycobacterium tuberculosis. Nat. Commun. 2023, 14, 1431.
- Yang, C.; Yuk, J.; Jo, E. The role of nitric oxide in mycobacterial infections. Immune Netw. 2009, 9, 46.
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brożek, J.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195.
- Marx, G.E.; Chan, E.D. Tuberculous meningitis: Diagnosis and treatment Overview. Tuberc. Res. Treat. 2011, 2011, 798764.
- Davis, A.; Meintjes, G.; Wilkinson, R.J. Treatment of tuberculous meningitis and its complications in adults. Curr. Treat. Options Neurol. 2018, 20, 5.
- Thwaites, G.; Hien, T.T. Tuberculous meningitis: Many questions, too few answers. Lancet Neurol. 2005, 4, 160–170.
- Kumar, R.; Kolloli, A.; Singh, P.; Vinnard, C.; Kaplan, G.; Subbian, S. Thalidomide and phosphodiesterase 4 inhibitors as host directed therapeutics for tuberculous Meningitis: Insights from the Rabbit Model. Front. Cell. Infect. Microbiol. 2020, 9, 450.
- Thwaites, G.; Bang, N.D.; Dung, N.H.; Quy, H.T.; Oanh, D.T.T.; Thoa, N.T.C.; Hien, N.Q.; Thuc, N.T.; Hải, N.N.; Lan, N.T.N.; et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N. Engl. J. Med. 2004, 351, 1741–1751.
- Davis, A.; Donovan, J.; Bremer, M.; Van Toorn, R.; Schoeman, J.; Dadabhoy, A.; Lai, R.; Cresswell, F.; Boulware, D.R.; Wilkinson, R.J.; et al. Host directed therapies for tuberculous meningitis. Wellcome Open Res. 2021, 5, 292.
- Ren, J.; Li, C.; Yan, X.; Qu, Y.; Yang, Y.; Guo, Z. Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms. Oxidative Med. Cell. Longev. 2021, 2021, 6643382.
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron chelators in treatment of iron overload. J. Toxicol. 2022, 2022, 4911205.
- Zhang, S.; Wei, X.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40.
- Abedi, A.; Ghobadi, H.; Sharghi, A.; Iranpour, S.; Fazlzadeh, M.; Aslani, M.R. Effect of saffron supplementation on oxidative stress markers (MDA, TAC, TOS, GPx, SOD, and pro-oxidant/antioxidant balance): An updated systematic review and meta-analysis of randomized placebo-controlled trials. Front. Med. 2023, 10, 1071514.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
671
Revision:
1 time
(View History)
Update Date:
07 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No