Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Devid Damiani | -- | 8022 | 2024-03-05 10:49:17 | | | |
| 2 | Catherine Yang | -2 word(s) | 8020 | 2024-03-06 02:21:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Damiani, D.; Baggiani, M.; Della Vecchia, S.; Naef, V.; Santorelli, F.M. HSP-Related iPS Cell Lines. Encyclopedia. Available online: https://encyclopedia.pub/entry/55865 (accessed on 25 June 2026).
Damiani D, Baggiani M, Della Vecchia S, Naef V, Santorelli FM. HSP-Related iPS Cell Lines. Encyclopedia. Available at: https://encyclopedia.pub/entry/55865. Accessed June 25, 2026.
Damiani, Devid, Matteo Baggiani, Stefania Della Vecchia, Valentina Naef, Filippo Maria Santorelli. "HSP-Related iPS Cell Lines" Encyclopedia, https://encyclopedia.pub/entry/55865 (accessed June 25, 2026).
Damiani, D., Baggiani, M., Della Vecchia, S., Naef, V., & Santorelli, F.M. (2024, March 05). HSP-Related iPS Cell Lines. In Encyclopedia. https://encyclopedia.pub/entry/55865
Damiani, Devid, et al. "HSP-Related iPS Cell Lines." Encyclopedia. Web. 05 March, 2024.
Copy Citation
Hereditary spastic paraplegias (HSPs) comprise a family of degenerative diseases mostly hitting descending axons of corticospinal neurons. Depending on the gene and mutation involved, the disease could present as a pure form with limb spasticity, or a complex form associated with cerebellar and/or cortical signs such as ataxia, dysarthria, epilepsy, and intellectual disability. The progressive nature of HSPs invariably leads patients to require walking canes or wheelchairs over time. The advent of induced pluripotent stem (iPS) cells allowed instead the direct study of morphological and molecular properties of the patient’s affected neurons generated upon in vitro differentiation.
hereditary spastic paraplegia
induced pluripotent stem cells
pluripotent cells
1. SPG3A
Spastic paraplegia 3A (SPG3A) represents the most common cause of early-onset AD-HSP, with an average onset at the age of four years [1]. The disease generally occurs in a pure form with slow progressive spasticity and weakness in the lower limbs [2]. However, it can also manifest as a complicated form, in which spasticity is associated with peripheral neuropathy and distal amyotrophy because of lower motor neuron involvement [3]. Magnetic resonance imaging (MRI) showed significant alterations in the transverse areas of the cervical and thoracic spinal cord [4] and the presence of a thin corpus callosum (TCC) [5]. SPG3A is caused by mutations in the ATL-1 gene, encoding the protein Atlastin-1, localized in the ER where it remodels lipid membranes [6], mediating homotypic fusion of tubules to form polygonal ER networks [7] (Figure 1).
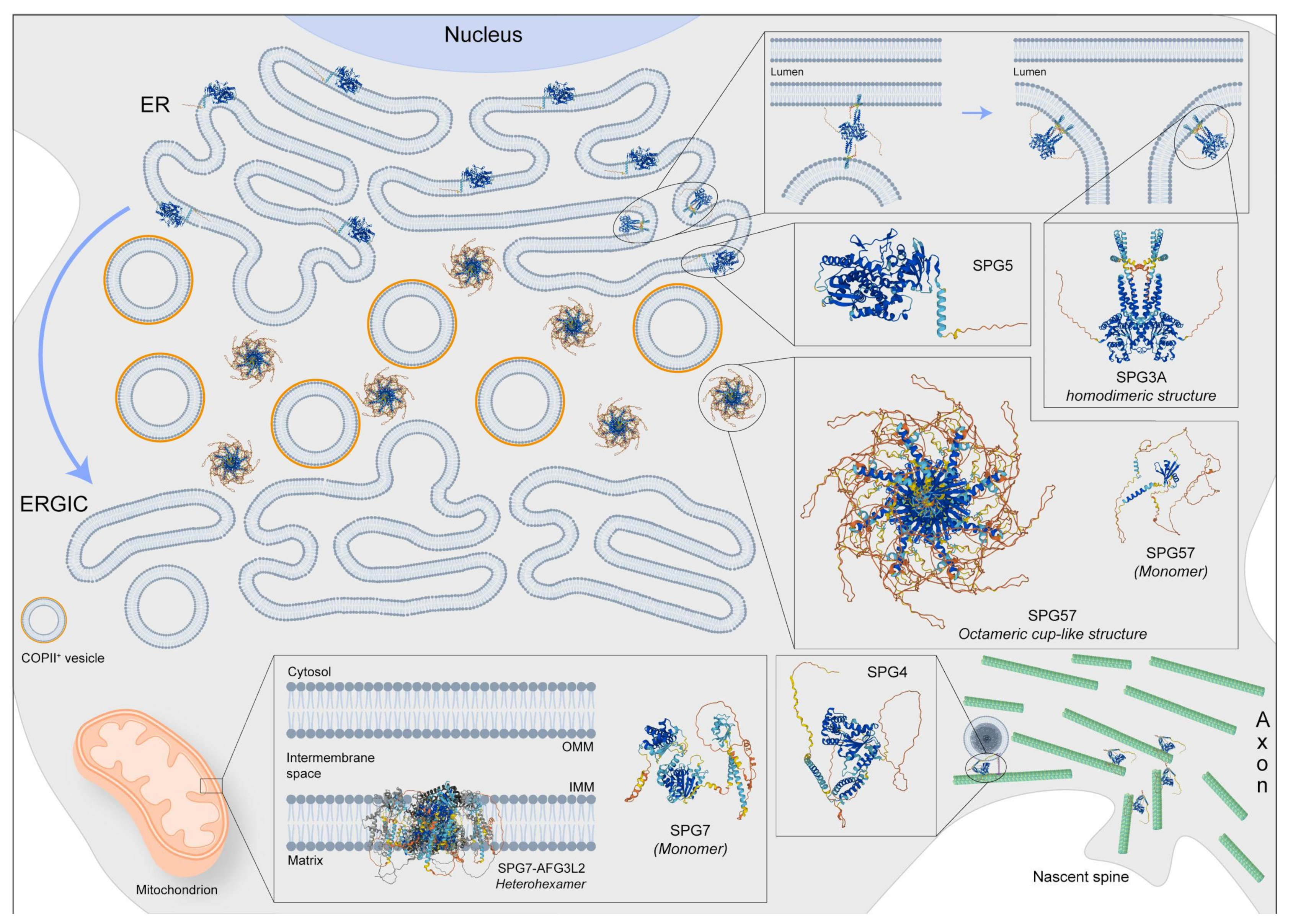
Figure 1. Representative image regarding the physiological localization and possible 3D structure of five SPG proteins: SPG3, SPG4, SPG5, SPG7, and SPG57. SPG3 is also involved in membrane fusion at the endoplasmic reticulum (ER), while SPG4 has several functions including microtubule dynamic in spine formation and regulation of vesicle transport. Moreover, the SGP5 protein should be localized at the ER but its function is poorly known in the brain. Instead, the SPG7 protein, forming the heterohexameric complex with AFG3L2, is an m-AAA protease, also regulating the degradation of damaged proteins, the synthesis respiratory chain, and the assembly of mitochondrial ribosomes. Finally, the SPG57 octameric structure is located between ER and ERGIC (ER-Golgi intermediate compartment) membranes, facilitating COPII vesicle export from the ER and retaining cargo-containing COPII vesicles at the ER/ERGIC interface. Protein 3D structures have been generated with Alphafold [8][9].
Disease-causing variants not only lead to altered vesicle trafficking between ER and Golgi but also to altered Golgi morphogenesis [10]. Although mutations in ATL-1 are predominantly missense, in-frame deletions have also been reported [11]. Together with Spastin, REEP1, and Reticulon 2, mutations in Atlastin-1 are responsible for abnormal membrane trafficking and abnormal organelle shaping; in fact, these proteins share similar hairpin loops that control ER membrane shaping.
In an effort to unveil the cellular and molecular bases of neuronal degeneration, several model systems have been generated. In mice, Atl-1 mRNA has been shown to be enriched in layer V of the cerebral cortex during early development [12]. There, Atlastin-1 is supposed to play an important role in the morphogenesis of dendrites [13], as knockdown in primary cortical neurons causes impairment of axonal growth and branching defects [14] (Figure 2A). Using episomal-mediated reprogramming, several pluripotent stem cell clones were derived from a 2-year-old female patient affected by pure HSP and carrying the heterozygous Pro342Ser mutation in ATL-1 [15], in a residue that is fully conserved across eukaryotes. Although the mutation did not abrogate protein translation and localization in patients’ fibroblasts, the mutated proline resides in a linker that is crucial for a conformational switch of Atlastin-1, strictly required for both the ER tubule fusion process and full GTPase activity. Similar to what had previously been observed in patient-derived fibroblasts, the Pro342Ser mutation did not affect Atlastin-1 protein levels in iPS-derived forebrain glutamatergic neurons. SPG3A cortical neurons derived from two different iPS clones displayed a significative reduction in the average length of axonal branches, implying an impairment of axonal outgrowth. In addition, in order to study the efficiency of anterograde and retrograde transport, mitochondrial movements across axonal compartments were monitored in iPS-derived mature (12 weeks old) cortical neurons by means of MitoTracker CMX Ros live-cell imaging. No significant differences were detected between mitochondrial transport velocities from control and SPG3A neurons, in both anterograde and retrograde directions, suggesting that the activity of molecular motors was unaffected by ATL-1 mutations. Despite this, a substantial decrease in anterograde motile events was detected in the SPG3A background, with only 50% of mitochondria actively moving in axons from mutant cells. Finally, as Atlastin-1 is an important binding partner of the SPG4 protein Spastin, a microtubule-severing AAA ATPase, researchers hypothesized that a microtubule-destabilizing approach could be beneficial in the SPG3A background. That said, administration of taxol and vinblastine was performed during SPG3A neuronal differentiation (Figure 2A), resulting in almost complete rescue of the axonal outgrowth phenotype in ATL-1 mutant cells and confirming the involvement of microtubule dynamics in SPG3A etiopathology.
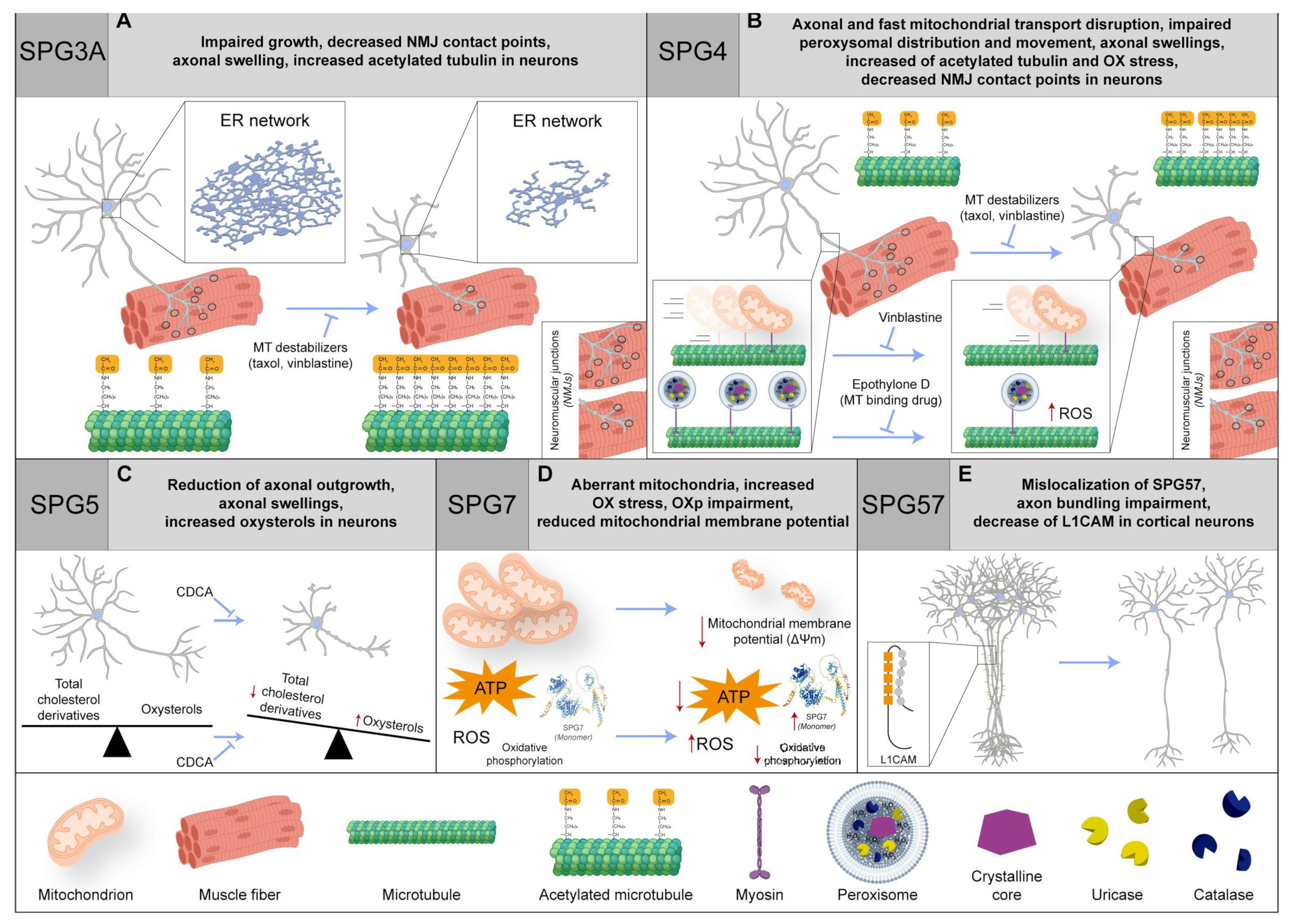
Figure 2. (A) In neurons, SPG3 impairment hampers the growth and NMJ contact points, causes axonal swelling, and increases the acetylated tubulin. Intriguingly, there is a recovery with MT-binding agents (such as taxol and vinblastine). (B) In SPG4 patients, the axonal transport is disrupted, coupled with an impairment in fast mitochondrial transport and peroxisome distribution and movement. The phenotype is rescued by vinblastine or epothilone D (MT-binding drug). Moreover, in neurons, axonal swelling is reported, and there is an increase in acetylated tubulin and oxidative (OX) stress, together with a decrease in NMJ contact points. Even in this case, a recovery with MT-binding agents (such as taxol and vinblastine) is found. (C) In SPG5 neurons, the axonal swelling phenomenon is found as well as an increase in oxysterols but not in total cholesterol derivatives and axonal outgrowth reduction. Importantly, chenodeoxycholic acid (CDCA) treatment can rescue the phenotype. (D) SPG7 patients with compound heterozygosity display an increased SPG7 quantity and OX stress, OX phosphorylation (OXp) impairment, and aberrant mitochondria, with a reduction in mitochondrial membrane potential and ATP production. (E) In SPG57 cortical neurons, there is an impairment of axon bundling, an L1CAM decrease, and SPG57 protein mislocalization. Red arrows indicate a dysregulation of biochemical pathways, unbalance of protein levels or changes in molecular specie concentrations. Protein 3D structures have been generated with Alphafold [8][9].
In another more recent study [16], iPS cells derived from three different SPG3A patients carrying heterozygous mutations in ATL-1 were differentiated in lower motoneurons following established protocols [17], generating a homogeneous population of cells expressing both the neuronal NEFH (Neurofilament Heavy) and ISL1 (Islet-1) motoneuron markers. Despite differentiation not being perturbed in the presence of mutated Atlastin-1, with no obviously detectable differences between the control and SPG3A cell lines, the immunostaining of patient-derived neurons revealed a significant increase in swelling in the axonal compartment, accompanied by a relative accumulation of acetylated tubulin (labeling stable microtubules; Figure 2A), compared to motoneurons derived from healthy iPSC lines. In addition, iPSCs were also differentiated in skeletal muscular tissue and combined with the cognate motoneurons in microfluidic devices, following established procedures [18] aimed at achieving in vitro formation of neuromuscular junctions (NMJs). The interaction of neurites with myotubes was confirmed in all cases, but the typical proportion of single- and multiple points of contact present in control NMJs was disrupted in SPG3A cell lines, with an increase in single-points and a decrease in multiple points of contact. Accordingly, a decrease in the expression for genes encoding acetylcholine receptor subunits (i.e., CHRNB1, CHRNA1, and CHRNG) was detected in SPG3A-mutated co-cultures, whereas no significant differences were found in genes related to inflammation (such as TNFa, IL6, IL1R, and IL10) or autophagy (e.g., BECN1, MAP1LC3A, MAP1LC3B, MAP1LC3C, and SQSTM1). Together, these experiments confirmed improper NMJ development in SPG3A disease and confirmed the usefulness of iPS cells for basic and preclinical research in this field.
2. SPG4
Spastic paraplegia 4 (SPG4; also known as SPAST-HSP) is the most frequent form in both familial and sporadic AD-HSPs [11][19]. Generally, patients have a pure phenotype with onset in teen years or early adulthood [20], but complicated features are also possible in SPG4 presentations [21], even with intra- and inter-familial variability [22][23]. Brain MRI is often normal or presenting with mild vermis atrophy, TCC, subtle white matter changes, and/or cerebellar atrophy [4]. Prominent spinal cord atrophy can also be retrieved at spinal cord MRI.
Patients affected by SPG4 carry mutations in SPAST. This gene codes for Spastin, a 616 amino acid (67.2 kDa) protein belonging to the AAA (ATPases associated with diverse cellular activities) group of proteins, involved in cell cycle regulation, protein degradation, organelle biogenesis, and vesicle-mediated functions [24] (Figure 1). Spastin has two isoforms, M1 and M87, differing in the presence or absence of 86 N-terminal amino acids, and each of them can be encoded by different isoforms lacking exon 4 (M1ΔEx4 and M87ΔEx4, respectively [25]). The protein is involved in microtubule disassembly [26] and enriched in the distal axon of corticospinal motoneurons [27], the nervous structure typically undergoing degeneration in HSP patients. Most pathogenic mutations involve the AAA domain and act through a mechanism of loss of function [28], predominantly causing axonal transport dysfunction [11] (Figure 2B). However, haploinsufficiency, or a combination of loss and gain of function, has also been invoked as a mutation-dependent mechanism for SPG4 [29].
To unravel the mechanisms underpinning the etiopathogenesis in SPG4, one of the first approaches consisted of deriving neural progenitor cells as floating neurospheres from biopsies of the SPG4 patient’s olfactory mucosa, successively propagated in adherent cultures as ONS (Olfactory NeuroSphere-derived) cells [30]. Similar models have been successfully used in other contexts to reveal significant and novel cellular aspects of genetically uncharacterized diseases such as schizophrenia and sporadic Parkinson’s disease. ONS cells derived from 9 patients and 10 healthy controls were compared via different approaches, such as flow cytometry, microarray-based gene expression profiling, and analysis of protein expression and cellular function. Strikingly, patient-derived cells gave homogeneous results for all experiments performed, irrespective of the type of SPAST mutation. In line with this, protein levels of both Spastin isoforms showed a 50% reduction in SPAST-mutated cells compared to wild type counterparts. In particular, gene expression was dysregulated, with differential expression for 57% of the genes analyzed. In line with the known function of the Spastin protein, cluster analysis of differentially expressed genes (DEGs) showed transcriptional regulation of genes regulating microtubule polymerization, stabilization, bundling, and organization, such as tubulins, kinesins, and Stathmin (encoded by STMN1 gene), most of them being overexpressed in SPAST-mutated cells. Consistently, the Stathmin protein level was increased by 50%, and this could partly explain why acetylated tubulin (a marker of stable microtubules), was decreased in ONS cells, contrary to expectations based on Spastin function (Figure 2B).
The same SPG4 patient- and healthy donor-derived ONS cell lines were used in a more recent study [31], where cells were differentiated into multipolar and bipolar cells containing axonal-like processes similar to neurites. As impairment of axonal transport is deemed a possible mechanism in SPG4 pathogenesis, the distribution of peroxisomes along processes was studied by staining neuron-like cells to label peroxisomal membrane protein PEX14. Differentiated SPG4-derived cells exhibited a substantial reduction in the peroxisome number in axon-like processes compared to control-derived neurons (58.06 vs. 80.4, respectively). In addition, differentiated ONS cells were transduced with a construct live-cell GFP peroxisome probe (CellLight Peroxisome-GFP BacMam 2.0, Thermo Scientific, Milan, Italy) to perform live imaging of moving peroxisomes along axon-like processes. Despite the overall motility behavior of single peroxisomes being conserved, suggesting normal interactions with microtubules and molecular motors, the percentage of fast-moving organelles was significantly reduced (2.3% vs. 10%) in SPG4 compared to the control population. In addition, axonal movements of peroxisomes shifted from retrograde to anterograde, generating a diverse cellular distribution that well correlates with increased sensitivity of SPAST mutant cells to physiological and peroxide-induced oxidative stress, as testified by increased immunoreactivity for the free radical compound 4-hydroxy-2-nonenal (4-HNE). Oxidative stress was completely rescued by treatment with the microtubule-stabilizing drug epothilone D (Figure 2B), corroborating cytoskeletal involvement in this phenotype, and raising the hypothesis that the impairment of peroxisome transport could create local domains of oxidative stress, ultimately leading to neuronal toxicity and cell death [32].
In another study [33], dermal fibroblasts taken from two patients carrying a heterozygous SPAST nonsense mutation served to generate iPS cells. Control fibroblast lines were derived from age-matched healthy controls with no history of movement disorder or neurologic disease. Spastin isoforms were evaluated in fibroblasts, iPSCs, NPCs, and primary astrocytes. While the two major isoforms present in every cell type were M87 and M87ΔEx4, the full-length M1 isoform was restricted exclusively to NPCs and neuronal cultures. Notably, the expression level of Spastin was increased in neural lineage. Spastin isoforms were overall downregulated in SPG4 iPS-derived neurons by nonsense-mediated decay, with a 34% and 40% decrease for M1 and M87 isoforms, respectively, corresponding with an overall decrease of almost 50% for the Spastin protein level. In addition, while the soma size did not change in the different experimental groups, SPG4 neurons presented significantly fewer primary neurites and reduced the total neuritic length, accompanied by a significant decrease in the number of branching points. Scholl analysis also confirmed the reduction in neuritic complexity in SPG4 neurons, especially in regions proximal to the cell soma. In addition, it is worth noting that the enlargement of axons for the focal accumulation of organelles, a condition termed axonal swelling [34][35], represents a common pathologic hallmark observed in the post-mortem spinal cord of SPG4 patients (Figure 2B). Differentiated neurons showed similar phenotypes, displaying disorganized and interrupted microtubules with abundant swellings and the accumulation of mitochondria. In spite of this, no differences were observed in terms of the total tubulin detyrosination or acetylation levels. A possible compensative mechanism in SPG4 cells could be represented by the observed upregulation of p60 Katanin, a protein with a similar microtubule-severing function as Spastin. As alteration of axonal transport is common in HSP, researchers investigated whether the altered morphology of the SPG4 neurites could also have an effect on organelle transport along axons. To do this, neurons were grown on microfluidic chambers, in a way that axonal projections could pass from one culture chamber to the other by crossing through the device’s microchannels [33]. Differentiated neurons were infected with lentiviruses expressing Mito-DsRed in order to visualize moving mitochondria in neurites via imaging of the microchannels. In this study, the amount of actively transported mitochondria was unchanged, similar to the speed of transported organelles. Despite this, imbalances in the anterograde and retrograde movements were identified (Figure 2B).
A similar study [36], reprogramming iPSC clones from patient fibroblasts carrying splice-site mutations, showed an increased abundance of acetylated tubulin upon differentiation in dorsal telencephalic neurons, confirming the hypothesis that the reduction in the Spastin level correlates with an increased quantity of stabilized microtubules. Most importantly, acetylated tubulin staining detected a substantial increase in swellings in axons of SPG4-derived neurons, especially in cells displaying longer neurites. As the accumulation of mitochondria (stained with MitoTracker Red CMXRos, Thermo Scientific, Milan, Italy) in swellings was also detected, it was possible to observe impaired fast axonal transport in 8-week-old SPG4-derived neurons, indicating that the impairment of retrograde axonal transport could be a key player in SPG4 pathogenesis. Finally, the link between SPG4 with increased microtubule stability was confirmed via rescuing the neuronal phenotype by treating it with microtubule destabilizing drugs. In particular, the administration of vinblastine at nanomolar concentrations significantly reduced the number of axonal swellings in 8-week-old SPG4-derived or SPAST knockdown neurons [36], definitely confirming the involvement of microtubule impairment in SPG4 pathogenesis (Figure 2B). To summarize, similar to SPG3, the use of iPS cells successfully achieved modeling for SPG4 disease, faithfully reproducing the cellular hallmark of patient neurons. It will be interesting in the future to see possible applications of microtubule drugs, such as taxol and vinblastine in both SPG3A and SPG4 patients, even considering the known side effects of these popular drugs.
3. SPG5
The regulation of cholesterol and biliary acid metabolism is crucial for the development and maintenance of central nervous systems, and defects in cholesterol-associated pathways could potentially lead to neurological diseases, such as amyotrophic lateral sclerosis (ALS) [37][38]. For instance, defects in cholesterol derivative degradation are associated with severe conditions, such as HSP and cerebrotendinous xanthomatosis (CTX). In the acidic pathway of degradation, cholesterol-containing compounds are oxidized at side chains, producing oxysterols (25- or 27-hydroxycholesterol, 25- or 27-OHC), which are in turn α-hydroxylated by the cytochrome P450 Family 7 Subfamily B Member 1 (CYP7B1) [39] (Figure 1). The accumulation of oxysterols, due to biallelic loss of function mutations in CYP7B1, causes neuronal toxicity and impairs synaptic function [40][41] associated with spastic paraplegia 5 (SPG5) [42]. SPG5 is a rare subtype of AR-HSP that typically manifests with periventricular and subcortical white matter lesions on brain MRI [43]. To shed light on the mechanism of the corticospinal axonal degeneration characteristic of human disease, skin fibroblasts derived from healthy donors and SPG5 patients were reprogrammed into iPS cells [44]. SPG5 pluripotent cells were then differentiated in cortical tissue using established protocols, starting from stem cell aggregates, and passing through a neuroepithelial rosette stage to the final glutamatergic forebrain specification [45][46]. Consistent with the known metabolic defect observed in the absence of the CYP7B1 protein, the total cholesterol amount was not affected in SPG5 cortical neurons, as revealed by the intensity of Filipin staining, whereas immuno-enzymatic quantifications performed via ELISA detected a significant increase of 27-OHC levels. Despite differentiating capacity being indistinguishable from control counterparts, SPG5-derived cortical projection neurons (cortical layer V, stained with CTIP2) exhibited decreased axonal length, determined as limited axonal outgrowth in 36-day-old neurons upon dissociation and replating. In addition, SPG5 mature (3-month-old) cortical projection neurons showed a dramatic increase in neuritic swellings detectable via staining for the Tau protein. Importantly, the treatment of chenodeoxycholic acid (CDCA) demonstrated effectiveness in reducing axonal varicosities of iPS-derived neurons (Figure 2C). CDCA is a natural farnesoid X receptor (FXR) agonist that acts to suppress cholesterol and bile acid biosynthesis through feedback mechanisms and reduces cholesterol accumulation [47][48]. For this reason, CDCA is routinely administered in the clinical treatment of CTX patients to mitigate symptoms and disease progression [49][50]. The findings obtained in that study were also corroborated by the coincident results observed in cortical projection neurons derived from isogenic CYP7B1 KO human embryonic stem cells, generated via CRISPR-Cas9-mediated genome editing. Overall, SPG5 iPSC-derived neurons proved to be a valid model faithfully recapitulating the hallmarks of HSP and providing a suitable platform for preclinical drug screening. This study showed that CDCA, a metabolic modulator of cholesterol, is a promising tool to counteract axonal neuropathy in SPG5 patients, while also counting the absence of drug tolerability and side effect issues due to common use in CTX management.
4. SPG7
Spastic paraplegia 7 (SPG7) is one of the most frequent AR-HSPs presenting a complicated phenotype that reproduces the multifaceted phenotype of mitochondrial diseases [51]. The disorder has a general adulthood onset and is characterized by leg weakness, spasticity, and other different manifestations, such as cerebellar signs/cerebellar atrophy, optic neuropathy [52][53][54], hearing loss, ptosis [55][56], and supranuclear palsy [57]. Hypokinetic movement disorders and lower motor neuron features can also be associated with SPG7.
SPG7 is caused by biallelic mutations in the SPG7 gene, encoding for Paraplegin, a zinc matrix metalloproteinase located in the inner mitochondrial membrane (Figure 1). Paraplegin is involved in multiple processes, such as protein quality control, proteolytic activation of essential mitochondrial proteins, ribosome assembly, and mitochondrial biogenesis. Ultrastructural studies performed in SPG7-derived cells have shown the occurrence of abnormally shaped mitochondria [52][58].
The previously mentioned ONS model proved useful to investigate mitochondrial dysfunction in SPG7 [59]. After staining with MitoTracker Green, SPG7 patient-derived ONS cells displayed short and highly fragmented mitochondria, with a low degree of branching and interconnectivity, which are typical signs of a dysfunctional mitochondrial network. Compared to the controls, SPG7 ONS displayed reduced basal respiration, significantly lower levels of oxygen consumption attributed to ATP production, maximal respiration, and reduced spare respiratory capacity. In addition, live cells were also stained with MitoSOX, a mitochondria-specific superoxide indicator, and CM-H2DCFDA, a general reactive oxygen species indicator. The observed fluorescence signals of both indicators were reported to be significantly higher in SPG7 mutant cells compared to the control counterparts (Figure 2D). Finally, SPG7 mutant cells displayed reduced cellular proliferation, in line with previously reported findings on cells carrying mutations in other known mitochondrial disease genes [60]. In addition, to study Paraplegin deficiency directly in cortical neurons, iPS cells were derived from peripheral blood mononuclear cells taken from SPG7 patients carrying different pathogenic variants [61]. Cells were differentiated in cortical progenitors and neurons via classical dual SMAD inhibition [62][63]. Despite the indistinguishable expression of pan-neuronal (beta-3 tubulin) and cortical (TBR1 and CTIP2) markers, neurite length and complexity were consistently reduced in patient-derived mature (30 days in vitro) cortical neurons compared to control cells. Moreover, both SPG7 progenitors and neurons displayed a slight increase in mitochondrial size-related parameters (i.e., whole area, perimeter, length, width), accompanied by a reduction in mitochondrial potential (as assessed via TMRM staining) and a likely correlated diminished cell viability. Transcriptomic analysis showed the downregulation of genes related to synaptic function in SPG7 neurons, and electrophysiological responses of SPG7 cortical neurons were also consistently reduced. As mitochondrial dysfunction was deemed to be the primal cause for all these phenotypes, differentiating SPG7 progenitors were treated with Bz-423, a drug shown to be effective in rescuing mitochondrial and neurological function in an SPG7 mouse model [64]. The treatment was able to rescue—almost completely—disease-associated neuronal phenotypes, posing hopes for the development of future therapeutic strategies in SPG7 patients, at least in the early stages of the disease. The same group obtained similar results using genuine patient-derived iPS-differentiated cortical neurons, which display reduced neuritic complexity, mitochondrial dysfunction, and increased degeneration. Also in this case, treatment with Bz-423 was able to rescue most of the phenotypes at nanomolar concentrations, thereby establishing a direct link between mitochondrial and neuronal defects in SPG7 neurons [61]. Unfortunately, Bz-423 induces cytostasis and cytotoxicity at higher concentrations and is immunomodulatory [65], so safer compounds will need to be tested as putative replacements. Still, SPG7 iPS-derived neurons will remain a very useful platform for drug discovery, as all studies discussed above certified these cells as a faithful model for SPG7.
5. SPG57
Biallelic mutations in TFG (tropomyosin receptor kinase) have been implicated in corticospinal axon pathology, causing neurological disorders, such as ALS, hereditary motor and sensory neuropathies (HMSNs), and Spastic paraplegia 57 (SPG57).
SPG57 presents in fewer families with lower limb spasticity, optic atrophy, and polyneuropathy. Consistently, the protein is highly expressed in mouse Purkinje cells and infragranular cortex, where it localizes either in the soma or axonal and dendritic projections [66][67][68][69].
TFG is a conserved regulator of the early secretory pathway, controlling the export of protein cargoes from ER. The protein multimerizes in cup-like octameric structures that create a meshwork between ER and ER-Golgi intermediate compartment (ERGIC), facilitating interaction between the scaffolding protein SEC-16 and COPII-coated transport carriers (Figure 1). In the absence of TFG, COP-II vesicles tend to scatter away ER exit sites, slowing down vesicle transport and triggering an ER stress response [70][71].
Overexpression of the Arg106Cys variant in cell lines not only disrupts normal ER organization but also alters the distribution of mitochondria, promoting clusters of these organelles around microtubule organizing centers. Despite this, cytoskeletal components themselves do not appear to be affected [67]. In agreement with these results, when overexpressed in mouse primary hippocampal neurons, known TFG pathogenic variants (Arg106Cys/His) dramatically increased mitochondrial fragmentation [72]. CRISPR-Cas9 genome editing in the well-characterized human IMRO90-4 iPS cell lines created an Arg106Cys model of TFG R106C [73]. The derived cortical glutamatergic neurons from control and genome-edited iPS cells revealed no differences in terms of timing of either neuronal differentiation or the emergence of spontaneous electrical activity. Also, the organization of Golgi was unaffected in mutant i-neurons, though the distribution of the variant protein itself was derailed, as staining was more diffuse and poorly localized at the ER/ERGIC interface. Different from findings in cultured mouse neurons, mitochondrial morphology, and motility along neurites were indistinguishable between control and mutant neurons, and there was neither impaired lysosomal function nor altered autophagic behavior. Finally, axons from neurons expressing TFG Arg106Cys did not show axonal swellings, a finding reproduced in other SPG iPS-derived neurons. To address instead the possibility of impaired axonal outgrowth, similar to SPG3A, telencephalic neurospheres were derived from expressing iPS cells. These floating aggregates, typically cultured on low adhesion surfaces, differentiate in neurons and start to send projections upon adhesion on coated coverslips. Strikingly, although no difference was detectable in terms of axonal outgrowth, neurites from SPG57-derived neurons failed to form axonal bundles, unlike what is usually observed in control neurons (Figure 2E). Homotypic axonal fasciculation is mostly due to the presence of transmembrane adhesion molecules that interact with the extracellular matrix with external domains [74]. A deeper investigation clarified that the defects lie in the early secretory pathway and impair trafficking of the adhesion molecule L1CAM, impairing the capacity of axons to organize in fascicles, a key factor for cells with extremely long projections like corticospinal motoneurons. Intriguingly, gene mutations in L1CAM have been associated with hydrocephalus, severe intellectual disability, aphasia, and HSP (SPG1) [75][76][77][78][79]. Induced pluripotent cells from SPG1 patients have been recently generated [80], and the characterization of differentiated cells is in progress. Also in this case, the use of iPS cells allowed phenotypic characterization of HSP neurons, defining axonal fasciculation issues in SPG57 cells and providing a platform for directed or unbiased pharmacological screening.
6. SPG11
Spastic paraplegia 11 (SPG11) is the most frequent AR-HSP, comprising about 20% of all AR-HSP and up to 45% of AR-HSP with TCC [51][81]. SPG11 patients generally have a complicated phenotype that includes progressive spasticity and weakness of the lower limbs, mild intellectual disability with learning difficulties, cerebellar signs, and peripheral neuropathy, usually associated with thinner corpus callosum at brain MRI [81][82]. It was also hypothesized that the phenotype results from combined degeneration of central and peripheral axons and neuronal loss within cortical, thalamic, and spinal cord regions [83].
SPG11 is caused by biallelic mutations in KIAA1840, leading to loss of protein function. The SPG11/KIAA1840 locus spans over 40 exons and encodes for a 2443-amino acid peptide named Spatacsin, a protein expressed ubiquitously in the nervous system, but most prominently in the cerebellum, cerebral cortex, and hippocampus [84][85]. In addition, Spatacsin is expressed throughout neural differentiation and present in dendrites and axons of mouse and human cortical projection neurons, where it associates mostly with synaptosomes. Although the precise function is unknown, Spatacsin has frequently been associated with vesicular trafficking and organelle shaping [11], and it has been associated with autophagy since its pivotal role in autophagic lysosome reformation (ALR) [86] (Figure 3). Downregulation of Spatacsin in vitro was performed via knockdown in primary cortical mouse neurons and the derivation of ES-differentiated cortical neurons with mutations in the SPG11 gene. In order to do this, human iPS cells were derived from three different patients carrying compound heterozygous mutations in SPG11. As controls, iPS cells were derived from two healthy Caucasian individuals with no history of movement disorder or neurological disease.
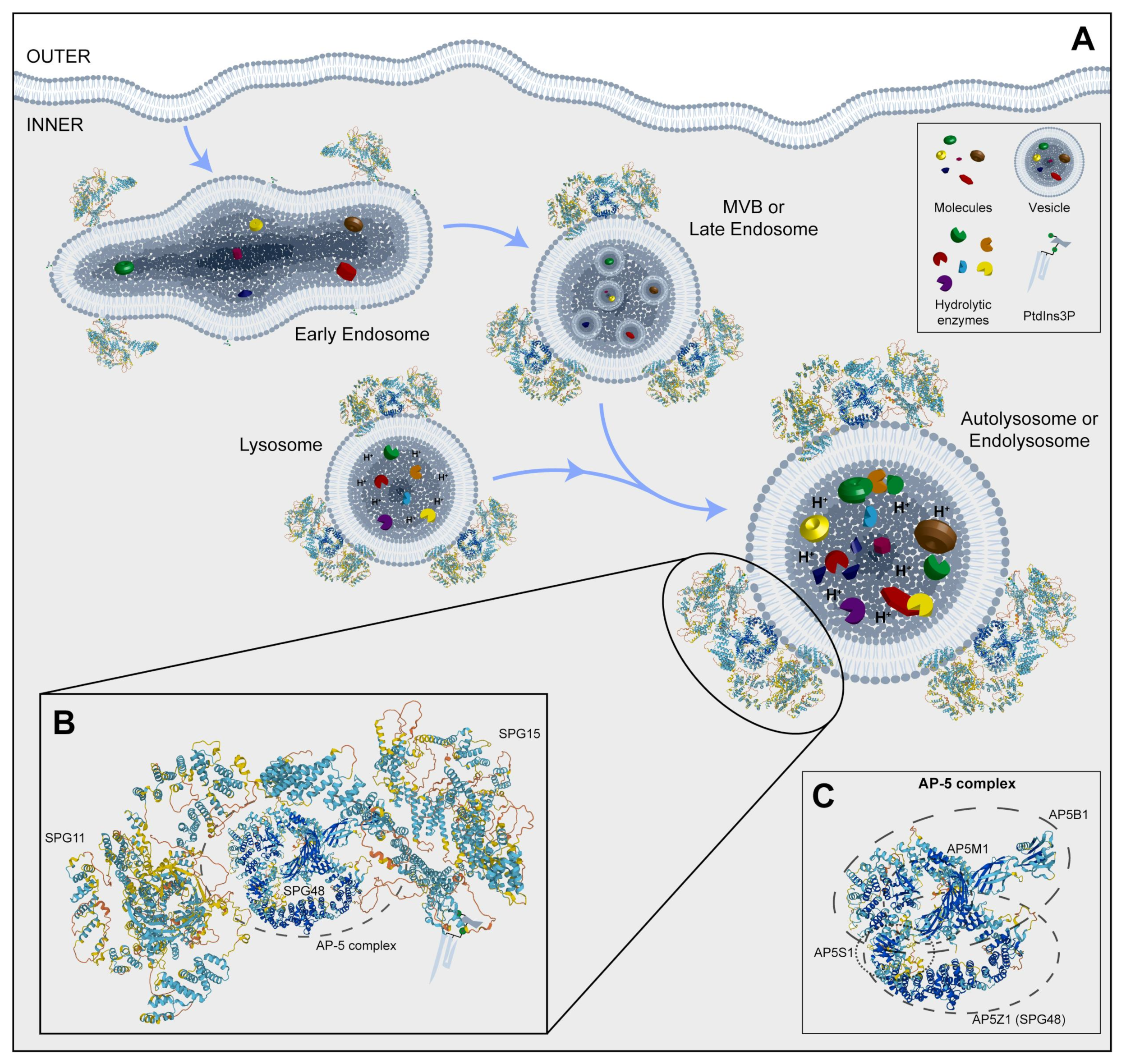
Figure 3. (A) Representative image of the endocytosis pathway and localization of SPG11, SPG15, and AP5Z1, which is a member of the AP-5 complex. (B) An inset of a possible 3D structure of the SPG11-SPG15-AP-5 complex on vesicles. (C) A box with a potential 3D structure of the AP-5 complex and its component four proteins, including SPG48. MVB: multivesicular body; PtdIns3P: phosphatidylinositol 3-phosphate. Protein 3D structures have been generated with Alphafold [8][9].
Spatacsin-depleted cortical neurons displayed a reduction in anterograde and an increase in retrograde axonal transport, accompanied by a reduction in acetylated tubulin signal, and it strictly correlated to neurite outgrowth impairment and a reduction in their complexity. Electron microscopy analysis of synaptic vesicles in SPG11 neurons demonstrated the presence of a large number of membrane-encircled inclusions within the neuritic compartment, while the expression analysis of transport-related genes pointed out the reduction in transcripts encoding anterograde molecular motors such as KIF3A, KIF5A, and KLC1, as well as tubulin associated genes (MAP, TAU, and TTBK1) and synaptic genes (VAMP2 and SYN1), Interestingly, Spatacsin knockdown not only interfered with axonal growth but also seemed to induce axonal retraction in primary mouse cortical neurons, well-correlating with axonal neuropathy found in SPG11 patients. These results were confirmed by time-lapse experiments studying the trafficking of synaptic vesicles in Synaptophysin-mCherry-transfected SPG11 cortical neurons cultured in microfluidic chambers. Consistently, mutant neurons displayed a significant reduction in anterograde transport, accompanied by an increased number of axonal processes showing no vesicular movement or even increased retrograde transport. Collectively, this scenario describes an efficient axonal transport reduction in SPG11 cortical neurons [87]. The effect of Spatacsin ablation was also studied in SPG11 iPS cell-derived spinal motoneurons [88]. Similar to cortical counterparts, differentiated cells displayed a reduction in the total neurite length, impaired mitochondrial anterograde axonal transport, and the presence of ultrastructural neuritic aggregates (Figure 4A–D). In addition, SPG11 motoneurons showed reduced mitochondrial membrane potential and increased lysosomal accumulation, consistent with SPG11 loss of function in animal models [89][90][91].
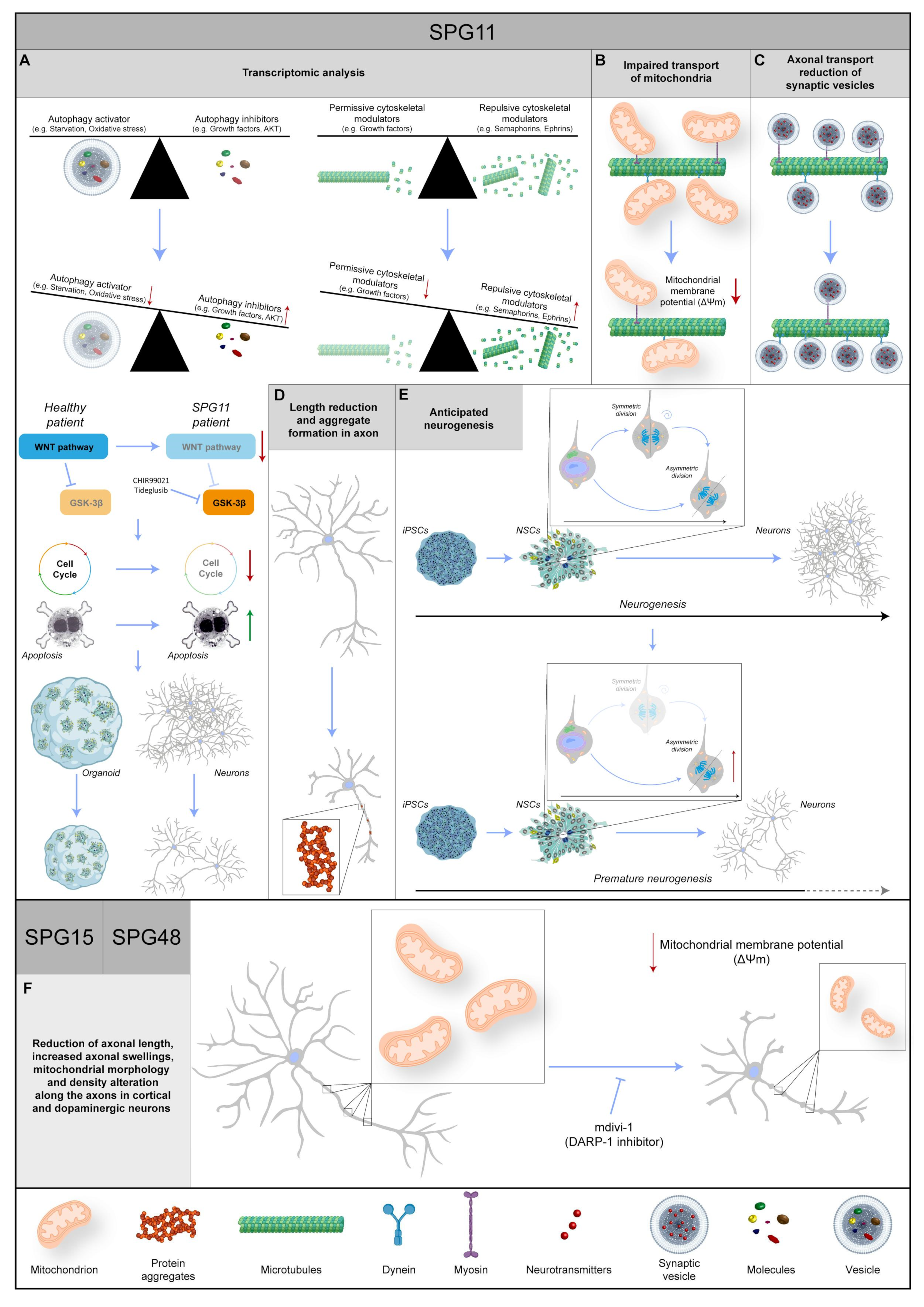
Figure 4. Representative images regarding the known data about three different SPG genes: SPG11, SPG15, and SPG48. (A) The transcriptomic analysis shows an increase in repulsive cytoskeletal modulators and autophagy inhibitors in combination with a decrease in permissive cytoskeletal modulators and autophagy activators. Moreover, in the SPG11 patient, the WNT pathway is impaired, leading to a reduction in proliferation and an increase in apoptosis, followed by a decrease in organoid size and the number of neurons. WNT pathway activation via inhibition of GSK-3β with CHIR99021 or Tideglusib rescues the phenotype. (B) In SPG11 neurons, the mitochondrial transport is impaired, and their membrane potential is reduced. (C) Reduction in synaptic vesicle axonal transport, in particular a decrease in anterograde and an increase in retrograde, is found in SPG11 patients. (D) In SPG11 motoneurons, the axons show a length reduction and the formation of aggregates. (E) The physiological timing of neurogenesis is disrupted in SPG11 patients, leading to premature differentiation, and resulting in a reduction in neuronal abundance. (F) In SPG15 and SPG48 patients, the axons in cortical and dopaminergic neurons show an increase in swelling phenomenon, a length reduction, and alterations in mitochondrial morphology and density. Intriguingly, most phenotypes are rescued with mdivi-1 (DARP-1 inhibitor). Red or green arrows indicate a dysregulation of biochemical pathways, unbalance of protein levels or changes in molecular specie concentrations. Protein 3D structures have been generated with Alphafold [8][9].
Gene ontology analysis of differentially regulated transcripts revealed over-represented biological processes mostly associated with distinct stages of neurodevelopmental pathways, including regulation of neurogenesis, nervous system development, and neuron differentiation. Interestingly, the KEGG pathway representation of individual sets of genes showed a clear downregulation of the Wnt pathway, cell cycle transcripts, and regulators of proliferation such as CCNA1 and CDH1, together with genes related to neuronal morphogenesis (Figure 4A). Mediators of repulsive neuronal interactions such as SEMA3A, EPHB1, PLXNB1, NFIA, and NCAM1 were found to instead be greatly upregulated. Those findings are also important since the impaired development of callosal fibers is a common finding in SPG11 patients [81]. Moreover, the expression of autophagy modulators was impaired, with the downregulation of positive and upregulation of negative regulators. Results from the transcriptomic analysis were corroborated with immunohistochemistry of hiPS-derived neural cells, showing reduced proliferation of neural progenitors and accelerated neurogenesis in the SPG11 background, ultimately leading to a reduction in the neuronal population. The formal demonstration of the involvement of the Wnt pathway was confirmed via the TCF/LEF (T-cell factor/lymphoid enhancer factor) ß-Catenin reporter, showing reduced luciferase activity in SPG11 cells, and definitely established by rescuing the observed cellular phenotype by forced activation of the cascade with CHIR99021 or Tideglusib [88]. Tideglusib is a known chemical inhibitor of GSK3 beta, the kinase that destabilizes ß-Catenin, presenting negative feedback to Wnt pathway activation (Figure 4A). Tideglusib treatment almost completely reverted the accumulation of axonal inclusions in Spatacsin-deficient iPS-derived cortical neurons, rescuing the effect on cell death [92].
Using the same SPG11 hiPS lines, the study on bidimensional culture was also extended to free-floating cerebral organoids, displaying a structural organization typical of developing cortical tissue. SPG11 organoids consistently presented smaller sizes in comparison to the control counterparts, with a significative reduction in the progenitor layer thickness after 9 weeks of culture and an enlarged volume of ventricle lumen. As the number of (neurogenic) vertical divisions outnumber horizontal (proliferative) divisions in the ventricular zone of SPG11 organoids, the phenotype observed seems to be caused by a mechanism of increased neurogenesis. Moreover, the cell cycle appears to be longer in Spatacsin-deficient cortical progenitors (Figure 4E). This collectively results in the generation of a consistently reduced number of cortical neurons in SPG11 cortical aggregates, faithfully mirroring what was previously observed in bidimensional cultures. Again, the modulation of the Wnt pathway with Tideglusib almost completely reverted the detrimental effects caused by the Spatacsin absence observed in SPG11 patient-derived cortical organoids (Figure 4A).
All these studies emphasized the role of Spatacsin in the generation and maintenance of cortical and spinal motoneurons, linking the development and neurodegeneration in SGP11 patients and opening new avenues for putative pharmacological treatments using Tideglusib, an FDA-approved drug for clinical use in dental care.
7. SPG15 and SPG48
Patients harboring variants in SPG15 or SPG48 forms of AR-HSP share similar clinical phenotypes to SPG11 patients, such as thinner corpus callosum, cognitive impairment, ataxia, cataracts, retinopathy, and early onset parkinsonism [82][93][94].
SPG15 is caused by biallelic mutations in ZFYVE26. Findings on brain MRI suggestive of SPG15 may include characteristic signal changes in the periventricular white matter, known as the “ears of the lynx” sign [95]. ZFYVE26 encodes for Spastizin, a protein described to be involved in endosomal trafficking, autophagy, and cytokinesis [93]. Mutated variants of Spastizin are indeed associated with defective autophagy [96]. On the other hand, SPG48 is a rare HSP presenting lower limb spasticity and associated with urinary incontinence caused by biallelic mutations in KIAA0415/AP5Z1. The gene encodes the AP5Z1 putative helicase, localized in both the nucleus and cytoplasm, and is involved in DNA double-strand break repair processes [97].
Spatacsin and Spastizin were shown to associate with the heterotetrameric adaptor protein complex 5 (AP-5), implicated in vesicle formation and sorting, while AP5Z1 protein is a subunit of the complex itself (Figure 3) [98]. Mouse models in which those genes have been knocked out consistently share a common impairment of lysosomal dynamics, with an accumulation of abnormal endolysosomes. According to the autosomal recessive nature of AP5Z1 mutations, fibroblasts derived from SPG48 patients contained no AP5Z1 protein. Interestingly, immunostaining of these cells for LAMP1, a known marker of late endosomes and lysosomes, showed the occurrence of numerous large positive puncta, and an accurate morphological examination performed via electron microscopy showed the presence of multilamellar structures filled with abnormal storage material forming exaggerated whorls, belts of striated material, fingerprint bodies, and some intraluminal vesicles [99]. Notably, similar ultrastructural findings have been reported for several lysosomal storage disorders, including metachromatic leukodystrophy, Fabry disease, mucopolysaccharidoses, Niemann–Pick disease, GM2 gangliosidoses, and neuronal ceroid lipofuscinosis [100][101][102], pointing to the hypothesis that AP-5 impairment leads to lysosomal accumulation. Interestingly, the absence of AP5Z1 in HeLa cells neither affected the total level of Spastizin nor the membrane association of Spatacsin. Conversely, the loss of Spatacsin or Spastizin resulted in decreased AP5Z1, suggesting a tight link between these proteins. As Spastizin and AP5Z1 are particularly enriched in neurons of the CNS [99], iPS cell lines were derived from SPG15 and SPG48 patients [103] in order to characterize SPG-derived differentiated neurons. Pluripotent cells were differentiated into cortical, spinal, and mesencephalic dopaminergic (mDA) neurons using established protocols [46][104][105][106] and characterized for the expression of their relative respective identity markers, such as Tbr1, HoxB4, and tyrosine hydroxylase (TH). Despite no difference being observed between SPG and control cells in terms of differentiation capacity for the different neuronal types, the ability to send projections upon neuronal detachment and replating was impaired in SPG15 and SPG48 cortical and dopaminergic neurons, but not spinal neurons. Analysis of neuritic outgrowth indeed showed that both the average neuritic length and the length of the longest neurite were found to be significantly reduced in patient-derived cells. In addition, the typical pathological hallmark of HSP neurons, namely axonal swelling, was present in 6-week-old SPG cortical neurons, as clearly stated by acetylated tubulin staining (Figure 4F).
A couple of premises have led researchers to pay attention to the mitochondrial structures and functions in SPG15 and SPG48. First, as aforementioned, both mutations can present as hypokinetic movement disorders like SPG11, and dopaminergic neurons seem to be particularly sensitive to the loss of function of mitochondrial proteins, such as PINK-1 and Parkin. Second, Spastizin was found to be localized at the mitochondrial surface [107]. Therefore, patient-derived neurons were stained with MitoTracker CMXRos to perform live imaging of mitochondrial morphology in the neuronal neuritic compartment. This experiment revealed a consistent reduction in the average mitochondrial length in both SPG15 and -48 cortical neurons, together with a reduction in the mitochondrial aspect ratio (defined as length/width) in SPG15 cells, a decreased number of mitochondria per 1 µm of neurite for the SPG48 neurons, and an overall reduction in linear mitochondrial density for both groups. Dopaminergic neurons also showed mitochondrial changes, with alterations in the length and aspect ratio in SPG15 neurons and a reduction in mitochondria along neurites in both SPG15 and SPG48 cell lines. In addition, together with morphology, the mitochondrial function of SPG-derived neurons was also assessed by measuring mitochondrial membrane potential with the fluorescent dye tetramethylrhodamine methyl ester (TMRM), as previously described [108]. These experiments revealed a significant reduction in mitochondrial membrane potential in both Spastizin and AP5Z1-deficient cortical neurons, pointing clearly to a decreased capacity of these cells to produce energy through respiratory pathways. In addition, this condition could facilitate the release of mitochondrial proteins in the cytoplasm, like AIF and cytochrome c, thereby triggering the mitochondrial pathway of the apoptotic mechanism [109]. Staining SPG15 and -48 long-term neuronal cultures with antibodies for activated Caspase-3 actually confirmed this hypothesis, with a dramatic increase in positive cells in the mutant neurons compared to the control counterparts. In the attempt to rescue these phenotypes, SPG neurons were treated with mdivi-1 (mitochondrial division inhibitor 1), an inhibitor of DRP1, a key protein mediating mitochondrial fission, as conducted in PINK-1 mutant mDA neurons [110]. Strikingly, a 48 h treatment with mdivi-1 reverted the reduction in the mitochondrial number in SPG telencephalic neurons, increasing their linear concentration in neurites, restoring their normal morphology, and decreasing apoptosis in telencephalic neurons (Figure 4F). Lentiviral knockdown of DRP1 in the same SPG cortical neurons confirmed a direct involvement of the protein in neuritic defects observed in SPG15 and SPG48 neurons. Notably, the same results obtained in SPG-derived telencephalic neurons were also reproduced with the shRNA-mediated knockdown of ZFYVE26 and AP5Z1 in H9 human embryonic stem cell-derived forebrain neurons [103]. These cells also displayed a reduction in neurite outgrowth, and similar to original SPG-derived neurons, the phenotype was rescued by treatment with DRP1 inhibitor mdivi-1.
Therefore, in the case of the SPG11-SPG15-SPG48 protein complex, the iPS cell-derived model offered a powerful tool to study mechanisms, corroborating their role in establishing and maintaining the intricate axonal projections in forebrain glutamatergic neurons. It will indeed be interesting in the future to see the possible utilization of mdivi-1 in clinical trials for these HSP forms.
8. AP-4 Complex (SPG47, SPG50, SPG51, SPG52)
Adaptor proteins (AP) constitute a conserved family of heterotrimeric protein complexes that work by facilitating the assembly of cargo proteins in vesicles and recruiting all the proteins required for transport and budding [111]. The AP-4 complex is composed of four different subunits, encoded by the genes AP4B1, AP4M1, AP4E1, and AP4S1 (Figure 5A). Biallelic mutations in these genes lead to typical forms of childhood-onset-complicated HSPs (SPG47, SPG50, SPG51, and SPG52, respectively), together called AP-4 complex-associated HSPs. Clinical manifestations in the AP-4 complex disease include delayed psychomotor development, progressive spasticity beginning in the lower limbs resulting over time in spastic tetraplegia, and intellectual disability with the absence of speech, post-natal microcephaly, and epilepsy [112]. Neuroimaging shows that TCC, nonspecific white matter abnormalities, ventriculomegaly, and colpocephaly can be observed, associated with cortical, cerebellar, and cerebral atrophy in patients with advanced disease.
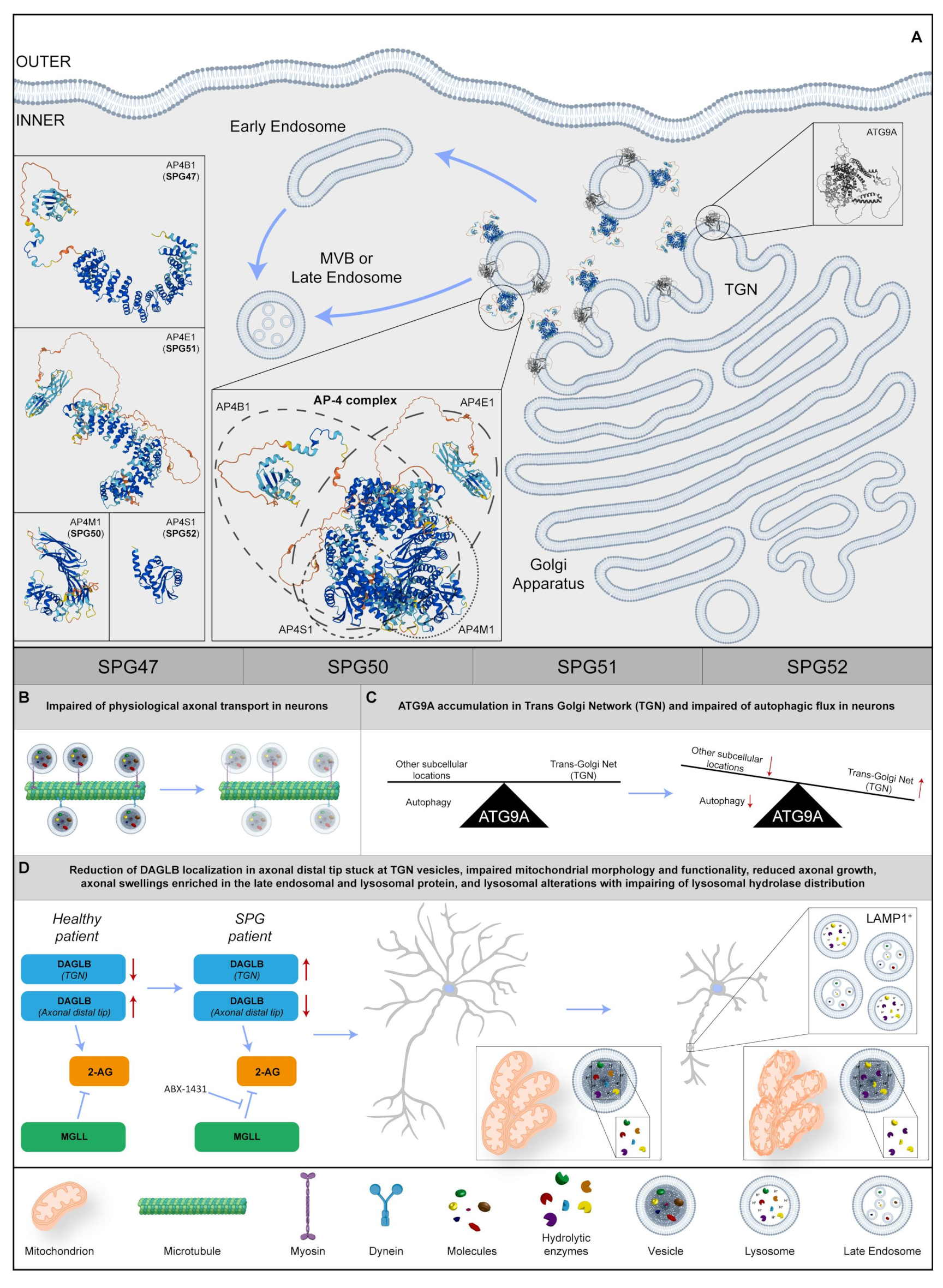
Figure 5. (A) Representative image regarding the physiological localization and possible 3D structure of four SPG proteins: SPG47, SPG50, SPG51, and SPG52. In particular, they shape the AP-4 complex at the Trans-Golgi Network (TGN) in the Golgi Apparatus, trafficking in transmembrane proteins from TGN to early or late endosomes. Regarding the insets on the left, 3D structures of each SPG protein are shown and, in the central box, there is a potential 3D structure of the AP-4 complex and the component’s four SPG proteins. (B) In the SPG patient, the axonal transport is disrupted in neurons. (C) In neurons, ATG9A remains at the TGN level, impairing the physiological autophagy. (D) In SPG neurons, DAGLB (diacylglycerol lipase-beta) accumulates in TGN, reducing 2-arachidonoylglycerol (2-AG) production in axonal tips. Inhibition of monoacylglycerol lipase (MGLL) rescues the axonal phenotype. Moreover, the impairment of axonal growth and axonal swelling is found, together with mitochondrial aberration and lysosomal dysfunction. Red arrows indicate a dysregulation of biochemical pathways, unbalance of protein levels or changes in molecular specie concentrations. Protein 3D structures have been generated with Alphafold [8][9].
The AP-4 complex is known to be implicated in the trafficking of transmembrane proteins, from TGN to early- and late-endosomal compartments [113]. Studies in cultured cells revealed that AP-4 is also involved in the transport of ATG9A, an important player in the formation of autophagosomes. The loss of the AP-4 function leads to the lack of the export of ATG9A from TGN, impairing its physiological axonal transport and proper neuronal autophagic flux [114][115] (Figure 5A–C). In line with these findings, the Ap4e1 knockout mouse displays reduced axonal developmental growth and the occurrence of axonal swellings [116][117].
To broaden the knowledge on AP-4 deficiency in human neurons, dermal fibroblast lines from seven patients carrying biallelic mutations in AP4B1, three in AP4M1, one in AP4E1, and three patients with mutations in AP4S1, were studied [118] to investigate levels and localization of ATG9A. The protein increased in patient fibroblasts, and almost completely colocalized with the trans-Golgi marker, TGN46. The rescue of the phenotype with the redistribution of ATG9A to the physiological subcellular localization was conducted by infecting AP4B1 fibroblasts with the lentiviral vector expressing AP4B1. AP4B1 fibroblasts were also used to derive iPS cells, and in turn, were differentiated in cortical glutamatergic neurons via the overexpression of Neurogenin-2 [119]. Induced neurons were harvested in 96-well plates and images were acquired through automated high-content confocal microscopy. With this approach it was possible to demonstrate that patient-derived i-neurons reproduced TGN accumulation of ATG9A, making this cellular phenotype a likely target for high-content drug screening tools. In addition, i-neurons showed impaired mitochondrial morphology and functionality (Figure 5D). In line with these results, morphological analysis of SPG51 patient-derived cortical neurons showed defective neuritic growth, mimicking in vitro the clinical findings of thin corpus callosum usually found in patients, and corroborating the hypothesis that mutant iPS-derived neurons could represent a faithful in vitro model to study pathological mechanisms of AP-4 HSP.
A more recent study exploited advanced proteomic approaches to search for cargo proteins of the AP-4 vesicle pathway, identifying DAGLB (diacylglycerol lipase-beta) as a novel interactor of the complex [120]. DAGLB is a serine lipase that hydrolyzes diacylglycerol (DAG) for the generation of 2-arachidonoylglycerol (2-AG), the most abundant endocannabinoid in the whole brain. The enzyme is localized at developmental stages in the axonal distal tip [121], where 2-AG is required to activate CB1 and CB2 cannabinoid receptors for axonal growth and guidance, especially for the elongation and fasciculation of the long axons of pyramidal cells [122][123][124]. Similar to what was observed for ATG9A, DAGLB showed a TGN subcellular localization in AP-4-deficient cell lines, and colocalization with the AP-4 accessory protein RUSC2 and cargo proteins SERINC1 and SERINC3 [120]. Most importantly, DAGLB localization was significantly reduced in axonal processes of AP4B1 mutant iPSC-derived neurons, with most of the protein stuck in TGN vesicles. As a most likely consequence, mutant neurons also showed a reduction in axonal length and branching, reminiscent of axonal pathology classically described in AP-4-HSP patients. As levels of 2-AG showed a considerable decrease in the brains of Ap-4-deficient mice, researchers elaborated a strategy to increase endocannabinoid levels via the chemical inhibition of monoacylglycerol lipase (MGLL, aka MAGL), the enzyme responsible for the hydrolysis of 2-AG into arachidonic acid [125]. Strikingly, treatment with the highly selective MGLL inhibitor (ABX-1431) was sufficient to rescue the axonal phenotype in AP-4-deficient neurons, while leaving control neurons completely unaffected. Taken together, these data strongly support the involvement of 2-AG in AP-4-HSP, opening the door for future therapeutic approaches with inhibitors of endocannabinoid catabolism (Figure 5D).
While autophagy involvement has been extensively studied in the pathophysiology of the AP-4 complex, few studies have characterized possible alterations of the lysosomal compartment in animal and cellular models. Interestingly, axonal swellings in the hippocampus and midbrain of Ap4e1 KO mice were enriched in the late endosomal and lysosomal protein, LAMP1 [126] (Figure 5D).
To fill this gap, multiple research groups [127][128][129] have made use of a CRISPRi-iPSC system to enable the Neurogenin-2-induced rapid generation of glutamatergic telencephalic neurons from pluripotent stem cells with knockdown of the AP4E1 gene.
The efficiency of the lysosomal proteolytic function, as assessed by the DQ-Red bovine serum albumin (BSA) trafficking assay, was decreased in mutant neurons, and the distribution of lysosomal hydrolases was also impaired in AP-4-deficient cells, suggesting that the protein complex regulates trafficking of specific lysosomal proteases (Figure 5D). As this process was known to be regulated by receptors such as Sortilin [130], it was possible to define the involvement of the lysosomal dysfunction in the pathogenesis of spastic paraplegia diseases due to AP-4 deficiency, thereby confirming the usefulness of patient-derived iPS cells in the identification of new potential molecular mechanisms.
9. Other SPG Genes
In addition to the aforementioned examples in common forms of HSP, a series of iPS cell lines have been derived from patients affected by other, relatively less common forms, including SPG1 [80], SPG10 [131], SPG30 [132], SPG43 [133], SPG56 [134], SPG58 [135], and SPG76 [136]. For all of these, extensive studies that characterize the differentiation of pluripotent cells in neuronal populations involved in the disease, such as cerebellar, dopaminergic, spinal, and corticospinal neurons, are needed.
Among them, SPG10, SPG30 and SPG58 are complicated forms of HSP caused by mutations in genes (KIF5A, KIF1A, and KIF1C, respectively) coding for kinesin molecular motors, critical for a proper synaptic function, especially for corticospinal motoneurons typically affected in paraparesis. KIF1A seems to be required for neurofilament transport [137], as well as for synaptic vesicle precursors [138][139] in mouse primary cortical and hippocampal neurons. Thus, the disruption of the kinesin function or even haploinsufficiency (as in SPG10 and SPG30) is sufficient enough to cause spastic symptoms [32][140].
On the other hand, SPG56 patients suffer from early-onset HSP with a wide spectrum of neurodevelopmental neuroimaging manifestations, including basal ganglia calcification, TCC, and hypomyelination [141][142]. The disease has been associated with null mutations in the CYP2U1 gene, coding for a member of the cytochrome P450 family, well-known for acting in the conversion of natural substrates into locally active signaling molecules. The CYP2U1 protein has been shown to participate in the hydrolyzation of riboflavin (vitamin B2), a flavonoid precursor of active compounds like flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), which are crucial for oxidative phosphorylation reactions in mitochondria. In addition, lipidomic studies in Cyp2u1 null brains imply high concentrations of coenzyme Q10 in patients. These data suggest that the reduction in ubiquinol in ubiquinone exerted by the FAD/FMN-dependent CoQ oxidoreductases might not work properly in SPG56 patients, with the obvious consequence of impaired cellular respiration particularly affecting sensitive cellular populations, such as corticospinal motoneurons and cone photoreceptors [142].
Finally, another example is SPG83, causing symptoms ranging from microcephaly and severe intellectual disability to pure late-onset HSP caused by biallelic variants in HPDL [143][144][145]. The gene encodes for an enzyme that was recently associated with an alternative pathway to coenzyme Q10 biosynthesis [146]. Our laboratory is directly involved in the derivation of several patient-derived pluripotent cell lines for the latter two forms.
References
- Hedera, P.; Fenichel, G.M.; Blair, M.; Haines, J.L. Novel Mutation in the SPG3A Gene in an African American Family with an Early Onset of Hereditary Spastic Paraplegia. Arch. Neurol. 2004, 61, 1600.
- Wilkinson, P.A.; Hart, P.E.; Patel, H.; Warner, T.T.; Crosby, A.H. SPG3A Mutation Screening in English Families with Early Onset Autosomal Dominant Hereditary Spastic Paraplegia. J. Neurol. Sci. 2003, 216, 43–45.
- Alecu, J.E.; Saffari, A.; Jordan, C.; Srivastava, S.; Blackstone, C.; Ebrahimi-Fakhari, D. De Novo Variants Cause Complex Symptoms in HSP- ATL1 (SPG3A) and Uncover Genotype–Phenotype Correlations. Hum. Mol. Genet. 2023, 32, 93–103.
- Da Graça, F.F.; de Rezende, T.J.R.; Vasconcellos, L.F.R.; Pedroso, J.L.; Barsottini, O.G.P.; França, M.C. Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives. Front. Neurol. 2019, 9, 1117.
- Orlacchio, A.; Montieri, P.; Babalini, C.; Gaudiello, F.; Bernardi, G.; Kawarai, T. Late-Onset Hereditary Spastic Paraplegia with Thin Corpus Callosum Caused by a New SPG3A Mutation. J. Neurol. 2011, 258, 1361–1363.
- Muriel, M.; Dauphin, A.; Namekawa, M.; Gervais, A.; Brice, A.; Ruberg, M. Atlastin-1, the Dynamin-like GTPase Responsible for Spastic Paraplegia SPG3A, Remodels Lipid Membranes and May Form Tubules and Vesicles in the Endoplasmic Reticulum. J. Neurochem. 2009, 110, 1607–1616.
- Ulengin, I.; Park, J.J.; Lee, T.H. ER Network Formation and Membrane Fusion by Atlastin1/SPG3A Disease Variants. Mol. Biol. Cell 2015, 26, 1616–1628.
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589.
- Namekawa, M.; Muriel, M.-P.; Janer, A.; Latouche, M.; Dauphin, A.; Debeir, T.; Martin, E.; Duyckaerts, C.; Prigent, A.; Depienne, C.; et al. Mutations in the SPG3A Gene Encoding the GTPase Atlastin Interfere with Vesicle Trafficking in the ER/Golgi Interface and Golgi Morphogenesis. Mol. Cell. Neurosci. 2007, 35, 1–13.
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary Spastic Paraplegia: Clinical-Genetic Characteristics and Evolving Molecular Mechanisms. Exp. Neurol. 2014, 261, 518–539.
- Shih, Y.-T.; Hsueh, Y.-P. VCP and ATL1 Regulate Endoplasmic Reticulum and Protein Synthesis for Dendritic Spine Formation. Nat. Commun. 2016, 7, 11020.
- Gao, Y.; Jiang, T.; Qu, C.; Tao, H.; Cao, H.; Zhao, Y.; Wang, Y.; Qu, J.; Chen, J.-G. Atlastin-1 Regulates Dendritic Morphogenesis in Mouse Cerebral Cortex. Neurosci. Res. 2013, 77, 137–142.
- Zhu, P.-P.; Soderblom, C.; Tao-Cheng, J.-H.; Stadler, J.; Blackstone, C. SPG3A Protein Atlastin-1 Is Enriched in Growth Cones and Promotes Axon Elongation during Neuronal Development. Hum. Mol. Genet. 2006, 15, 1343–1353.
- Zhu, P.-P.; Denton, K.R.; Pierson, T.M.; Li, X.-J.; Blackstone, C. Pharmacologic Rescue of Axon Growth Defects in a Human IPSC Model of Hereditary Spastic Paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648.
- Costamagna, D.; Casters, V.; Beltrà, M.; Sampaolesi, M.; Van Campenhout, A.; Ortibus, E.; Desloovere, K.; Duelen, R. Autologous IPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia. Cells 2022, 11, 3351.
- Bianchi, F.; Malboubi, M.; Li, Y.; George, J.H.; Jerusalem, A.; Szele, F.; Thompson, M.S.; Ye, H. Rapid and Efficient Differentiation of Functional Motor Neurons from Human IPSC for Neural Injury Modelling. Stem Cell Res. 2018, 32, 126–134.
- Paranjape, S.R.; Nagendran, T.; Poole, V.; Harris, J.; Taylor, A.M. Compartmentalization of Human Stem Cell-Derived Neurons within Pre-Assembled Plastic Microfluidic Chips. J. Vis. Exp. 2019, e59250.
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183.
- Parodi, L.; Fenu, S.; Barbier, M.; Banneau, G.; Duyckaerts, C.; Tezenas du Montcel, S.; Monin, M.-L.; Ait Said, S.; Guegan, J.; Tallaksen, C.M.E.; et al. Spastic Paraplegia Due to SPAST Mutations Is Modified by the Underlying Mutation and Sex. Brain 2018, 141, 3331–3342.
- Kumar, K.R.; Sue, C.M.; Burke, D.; Ng, K. Peripheral Neuropathy in Hereditary Spastic Paraplegia Due to Spastin (SPG4) Mutation—A Neurophysiological Study Using Excitability Techniques. Clin. Neurophysiol. 2012, 123, 1454–1459.
- Akaba, Y.; Takeguchi, R.; Tanaka, R.; Takahashi, S. A Complex Phenotype of a Patient with Spastic Paraplegia Type 4 Caused by a Novel Pathogenic Variant in the SPAST Gene. Case Rep. Neurol. 2021, 13, 763–771.
- Orlacchio, A.; Kawarai, T.; Totaro, A.; Errico, A.; St George-Hyslop, P.H.; Rugarli, E.I.; Bernardi, G. Hereditary Spastic Paraplegia: Clinical Genetic Study of 15 Families. Arch. Neurol. 2004, 61, 849–855.
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.-S.; Cruaud, C.; Dürr, A.; Wincker, P.; et al. Spastin, a New AAA Protein, Is Altered in the Most Frequent Form of Autosomal Dominant Spastic Paraplegia. Nat. Genet. 1999, 23, 296–303.
- Claudiani, P.; Riano, E.; Errico, A.; Andolfi, G.; Rugarli, E.I. Spastin Subcellular Localization Is Regulated through Usage of Different Translation Start Sites and Active Export from the Nucleus. Exp. Cell Res. 2005, 309, 358–369.
- Errico, A.; Ballabio, A.; Rugarli, E.I. Spastin, the Protein Mutated in Autosomal Dominant Hereditary Spastic Paraplegia, Is Involved in Microtubule Dynamics. Hum. Mol. Genet. 2002, 11, 153–163.
- Salinas, S.; Carazo-Salas, R.E.; Proukakis, C.; Schiavo, G.; Warner, T.T. Spastin and Microtubules: Functions in Health and Disease. J. Neurosci. Res. 2007, 85, 2778–2782.
- Beetz, C.; Nygren, A.O.H.; Schickel, J.; Auer-Grumbach, M.; Bürk, K.; Heide, G.; Kassubek, J.; Klimpe, S.; Klopstock, T.; Kreuz, F.; et al. High Frequency of Partial SPAST Deletions in Autosomal Dominant Hereditary Spastic Paraplegia. Neurology 2006, 67, 1926–1930.
- Piermarini, E.; Akarsu, S.; Connors, T.; Kneussel, M.; Lane, M.A.; Morfini, G.; Karabay, A.; Baas, P.W.; Qiang, L. Modeling Gain-of-Function and Loss-of-Function Components of SPAST-Based Hereditary Spastic Paraplegia Using Transgenic Mice. Hum. Mol. Genet. 2022, 31, 1844–1859.
- Abrahamsen, G.; Fan, Y.; Matigian, N.; Wali, G.; Bellette, B.; Sutharsan, R.; Raju, J.; Wood, S.A.; Veivers, D.; Sue, C.M.; et al. A Patient-Derived Stem Cell Model of Hereditary Spastic Paraplegia with SPAST Mutations. Dis. Model. Mech. 2013, 6, 489–502.
- Wali, G.; Sutharsan, R.; Fan, Y.; Stewart, R.; Tello Velasquez, J.; Sue, C.M.; Crane, D.I.; Mackay-Sim, A. Mechanism of Impaired Microtubule-Dependent Peroxisome Trafficking and Oxidative Stress in SPAST-Mutated Cells from Patients with Hereditary Spastic Paraplegia. Sci. Rep. 2016, 6, 27004.
- Crane, D.I. Revisiting the Neuropathogenesis of Zellweger Syndrome. Neurochem. Int. 2014, 69, 1–8.
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plotz, S.; Boyer, L.; Marchetto, M.C.N.; et al. Gene Dosage-Dependent Rescue of HSP Neurite Defects in SPG4 Patients’ Neurons. Hum. Mol. Genet. 2014, 23, 2527–2541.
- Tarrade, A.; Fassier, C.; Courageot, S.; Charvin, D.; Vitte, J.; Peris, L.; Thorel, A.; Mouisel, E.; Fonknechten, N.; Roblot, N.; et al. A Mutation of Spastin Is Responsible for Swellings and Impairment of Transport in a Region of Axon Characterized by Changes in Microtubule Composition. Hum. Mol. Genet. 2006, 15, 3544–3558.
- Kasher, P.R.; De Vos, K.J.; Wharton, S.B.; Manser, C.; Bennett, E.J.; Bingley, M.; Wood, J.D.; Milner, R.; McDermott, C.J.; Miller, C.C.J.; et al. Direct Evidence for Axonal Transport Defects in a Novel Mouse Model of Mutant Spastin-induced Hereditary Spastic Paraplegia (HSP) and Human HSP Patients. J. Neurochem. 2009, 110, 34–44.
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.-J. Loss of Spastin Function Results in Disease-Specific Axonal Defects in Human Pluripotent Stem Cell-Based Models of Hereditary Spastic Paraplegia. Stem Cells 2014, 32, 414–423.
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.-Å.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R.; et al. Defective Cholesterol Metabolism in Amyotrophic Lateral Sclerosis. J. Lipid Res. 2017, 58, 267–278.
- Rickman, O.J.; Baple, E.L.; Crosby, A.H. Lipid Metabolic Pathways Converge in Motor Neuron Degenerative Diseases. Brain 2020, 143, 1073–1087.
- Tsaousidou, M.K.; Ouahchi, K.; Warner, T.T.; Yang, Y.; Simpson, M.A.; Laing, N.G.; Wilkinson, P.A.; Madrid, R.E.; Patel, H.; Hentati, F.; et al. Sequence Alterations within CYP7B1 Implicate Defective Cholesterol Homeostasis in Motor-Neuron Degeneration. Am. J. Hum. Genet. 2008, 82, 510–515.
- Merino-Serrais, P.; Loera-Valencia, R.; Rodriguez-Rodriguez, P.; Parrado-Fernandez, C.; Ismail, M.A.; Maioli, S.; Matute, E.; Jimenez-Mateos, E.M.; Björkhem, I.; DeFelipe, J.; et al. 27-Hydroxycholesterol Induces Aberrant Morphology and Synaptic Dysfunction in Hippocampal Neurons. Cereb. Cortex 2019, 29, 429–446.
- Wang, Y.; An, Y.; Zhang, D.; Yu, H.; Zhang, X.; Wang, Y.; Tao, L.; Xiao, R. 27-Hydroxycholesterol Alters Synaptic Structural and Functional Plasticity in Hippocampal Neuronal Cultures. J. Neuropathol. Exp. Neurol. 2019, 78, 238–247.
- Schüle, R.; Siddique, T.; Deng, H.-X.; Yang, Y.; Donkervoort, S.; Hansson, M.; Madrid, R.E.; Siddique, N.; Schöls, L.; Björkhem, I. Marked Accumulation of 27-Hydroxycholesterol in SPG5 Patients with Hereditary Spastic Paresis. J. Lipid Res. 2010, 51, 819–823.
- Biancheri, R.; Ciccolella, M.; Rossi, A.; Tessa, A.; Cassandrini, D.; Minetti, C.; Santorelli, F.M. White Matter Lesions in Spastic Paraplegia with Mutations in SPG5/CYP7B1. Neuromuscul. Disord. 2009, 19, 62–65.
- Mou, Y.; Nandi, G.; Mukte, S.; Chai, E.; Chen, Z.; Nielsen, J.E.; Nielsen, T.T.; Criscuolo, C.; Blackstone, C.; Fraidakis, M.J.; et al. Chenodeoxycholic Acid Rescues Axonal Degeneration in Induced Pluripotent Stem Cell-Derived Neurons from Spastic Paraplegia Type 5 and Cerebrotendinous Xanthomatosis Patients. Orphanet J. Rare Dis. 2023, 18, 72.
- Boisvert, E.M.; Denton, K.; Lei, L.; Li, X.-J. The Specification of Telencephalic Glutamatergic Neurons from Human Pluripotent Stem Cells. J. Vis. Exp. 2013, e50321.
- Li, X.-J.; Zhang, X.; Johnson, M.A.; Wang, Z.-B.; LaVaute, T.; Zhang, S.-C. Coordination of Sonic Hedgehog and Wnt Signaling Determines Ventral and Dorsal Telencephalic Neuron Types from Human Embryonic Stem Cells. Development 2009, 136, 4055–4063.
- Bramlett, K.S.; Yao, S.; Burris, T.P. Correlation of Farnesoid X Receptor Coactivator Recruitment and Cholesterol 7α-Hydroxylase Gene Repression by Bile Acids. Mol. Genet. Metab. 2000, 71, 609–615.
- Kallner, M. The Effect of Chenodeoxycholic Acid Feeding on Bile Acid Kinetics and Fecal Neutral Steroid Excretion in Patients with Hyperlipoproteinemia Types II and IV. J. Lab. Clin. Med. 1975, 86, 595–604.
- Bonnot, O.; Fraidakis, M.J.; Lucanto, R.; Chauvin, D.; Kelley, N.; Plaza, M.; Dubourg, O.; Lyon-Caen, O.; Sedel, F.; Cohen, D. Cerebrotendinous Xanthomatosis Presenting with Severe Externalized Disorder: Improvement after One Year of Treatment with Chenodeoxycholic Acid. CNS Spectr. 2010, 15, 231–237.
- Dai, D.; Mills, P.B.; Footitt, E.; Gissen, P.; McClean, P.; Stahlschmidt, J.; Coupry, I.; Lavie, J.; Mochel, F.; Goizet, C.; et al. Liver Disease in Infancy Caused by Oxysterol 7α-hydroxylase Deficiency: Successful Treatment with Chenodeoxycholic Acid. J. Inherit. Metab. Dis. 2014, 37, 851–861.
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary Spastic Paraplegia: Clinicogenetic Lessons from 608 Patients. Ann. Neurol. 2016, 79, 646–658.
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic Paraplegia and OXPHOS Impairment Caused by Mutations in Paraplegin, a Nuclear-Encoded Mitochondrial Metalloprotease. Cell 1998, 93, 973–983.
- De Michele, G.; De Fusco, M.; Cavalcanti, F.; Filla, A.; Marconi, R.; Volpe, G.; Monticelli, A.; Ballabio, A.; Casari, G.; Cocozza, S. A New Locus for Autosomal Recessive Hereditary Spastic Paraplegia Maps to Chromosome 16q24.3. Am. J. Hum. Genet. 1998, 63, 135–139.
- McDermott, C.J.; Dayaratne, R.K.; Tomkins, J.; Lusher, M.E.; Lindsey, J.C.; Johnson, M.A.; Casari, G.; Turnbull, D.M.; Bushby, K.; Shaw, P.J. Paraplegin Gene Analysis in Hereditary Spastic Paraparesis (HSP) Pedigrees in Northeast England. Neurology 2001, 56, 467–471.
- Warnecke, T.; Duning, T.; Schirmacher, A.; Mohammadi, S.; Schwindt, W.; Lohmann, H.; Dziewas, R.; Deppe, M.; Ringelstein, E.B.; Young, P. A Novel Splice Site Mutation in the SPG7 Gene Causing Widespread Fiber Damage in Homozygous and Heterozygous Subjects. Mov. Disord. 2010, 25, 413–420.
- Tzoulis, C.; Denora, P.S.; Santorelli, F.M.; Bindoff, L.A. Hereditary Spastic Paraplegia Caused by the Novel Mutation 1047insC in the SPG7 Gene. J. Neurol. 2008, 255, 1142–1144.
- Warnecke, T.; Duning, T.; Schwan, A.; Lohmann, H.; Epplen, J.T.; Young, P. A Novel Form of Autosomal Recessive Hereditary Spastic Paraplegia Caused by a New SPG7 Mutation. Neurology 2007, 69, 368–375.
- Nolden, M.; Ehses, S.; Koppen, M.; Bernacchia, A.; Rugarli, E.I.; Langer, T. The M-AAA Protease Defective in Hereditary Spastic Paraplegia Controls Ribosome Assembly in Mitochondria. Cell 2005, 123, 277–289.
- Wali, G.; Kumar, K.R.; Liyanage, E.; Davis, R.L.; Mackay-Sim, A.; Sue, C.M. Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 2020, 14, 820.
- James, A.M.; Wei, Y.H.; Pang, C.Y.; Murphy, M.P. Altered Mitochondrial Function in Fibroblasts Containing MELAS or MERRF Mitochondrial DNA Mutations. Biochem. J. 1996, 318, 401–407.
- Wali, G.; Li, Y.; Liyanage, E.; Kumar, K.R.; Day, M.L.; Sue, C.M. Pharmacological Rescue of Mitochondrial and Neuronal Defects in SPG7 Hereditary Spastic Paraplegia Patient Neurons Using High Throughput Assays. Front. Neurosci. 2023, 17, 1231584.
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly Efficient Neural Conversion of Human ES and IPS Cells by Dual Inhibition of SMAD Signaling. Nat. Biotechnol. 2009, 27, 275–280.
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.-W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed Differentiation and Functional Maturation of Cortical Interneurons from Human Embryonic Stem Cells. Cell Stem Cell 2013, 12, 559–572.
- Sambri, I.; Massa, F.; Gullo, F.; Meneghini, S.; Cassina, L.; Carraro, M.; Dina, G.; Quattrini, A.; Patanella, L.; Carissimo, A.; et al. Impaired Flickering of the Permeability Transition Pore Causes SPG7 Spastic Paraplegia. EBioMedicine 2020, 61, 103050.
- Sundberg, T.B.; Ney, G.M.; Subramanian, C.; Opipari, A.W.; Glick, G.D. The Immunomodulatory Benzodiazepine Bz-423 Inhibits B-Cell Proliferation by Targeting c-Myc Protein for Rapid and Specific Degradation. Cancer Res. 2006, 66, 1775–1782.
- Ishiura, H.; Sako, W.; Yoshida, M.; Kawarai, T.; Tanabe, O.; Goto, J.; Takahashi, Y.; Date, H.; Mitsui, J.; Ahsan, B.; et al. The TRK-Fused Gene Is Mutated in Hereditary Motor and Sensory Neuropathy with Proximal Dominant Involvement. Am. J. Hum. Genet. 2012, 91, 320–329.
- Beetz, C.; Johnson, A.; Schuh, A.L.; Thakur, S.; Varga, R.-E.; Fothergill, T.; Hertel, N.; Bomba-Warczak, E.; Thiele, H.; Nürnberg, G.; et al. Inhibition of TFG Function Causes Hereditary Axon Degeneration by Impairing Endoplasmic Reticulum Structure. Proc. Natl. Acad. Sci. USA 2013, 110, 5091–5096.
- Kawarai, T.; Morita, M.; Morigaki, R.; Fujita, K.; Nodera, H.; Izumi, Y.; Goto, S.; Nakano, I.; Kaji, R. Pathomechanisms of Motor Neuron Death by Mutant TFG. Rinsho Shinkeigaku 2013, 53, 1199.
- Yagi, T.; Ito, D.; Suzuki, N. TFG-Related Neurologic Disorders: New Insights into Relationships between Endoplasmic Reticulum and Neurodegeneration. J. Neuropathol. Exp. Neurol. 2016, 75, 299–305.
- Witte, K.; Schuh, A.L.; Hegermann, J.; Sarkeshik, A.; Mayers, J.R.; Schwarze, K.; Yates III, J.R.; Eimer, S.; Audhya, A. TFG-1 Function in Protein Secretion and Oncogenesis. Nat. Cell Biol. 2011, 13, 550–558.
- Johnson, A.; Bhattacharya, N.; Hanna, M.; Pennington, J.G.; Schuh, A.L.; Wang, L.; Otegui, M.S.; Stagg, S.M.; Audhya, A. TFG Clusters COPII-coated Transport Carriers and Promotes Early Secretory Pathway Organization. EMBO J. 2015, 34, 811–827.
- Harlalka, G.V.; McEntagart, M.E.; Gupta, N.; Skrzypiec, A.E.; Mucha, M.W.; Chioza, B.A.; Simpson, M.A.; Sreekantan-Nair, A.; Pereira, A.; Günther, S.; et al. Novel Genetic, Clinical, and Pathomechanistic Insights into TFG-Associated Hereditary Spastic Paraplegia. Hum. Mutat. 2016, 37, 1157–1161.
- Slosarek, E.L.; Schuh, A.L.; Pustova, I.; Johnson, A.; Bird, J.; Johnson, M.; Frankel, E.B.; Bhattacharya, N.; Hanna, M.G.; Burke, J.E.; et al. Pathogenic TFG Mutations Underlying Hereditary Spastic Paraplegia Impair Secretory Protein Trafficking and Axon Fasciculation. Cell Rep. 2018, 24, 2248–2260.
- Winther, M.; Walmod, P.S. Neural Cell Adhesion Molecules Belonging to the Family of Leucine-Rich Repeat Proteins. Adv. Neurobiol. 2014, 8, 315–395.
- Jouet, M.; Rosenthal, A.; Armstrong, G.; MacFarlane, J.; Stevenson, R.; Paterson, J.; Metzenberg, A.; Ionasescu, V.; Temple, K.; Kenwrick, S. X–Linked Spastic Paraplegia (SPG1), MASA Syndrome and X–Linked Hydrocephalus Result from Mutations in the L1 Gene. Nat. Genet. 1994, 7, 402–407.
- Rosenthal, A.; Jouet, M.; Kenwrick, S. Aberrant Splicing of Neural Cell Adhesion Molecule L1 MRNA in a Family with X-Linked Hydrocephalus. Nat. Genet. 1992, 2, 107–112.
- Stumpel, C.; Vos, Y.J. L1 Syndrome; University of Washington: Seattle, WA, USA, 1993.
- Vos, Y.J.; de Walle, H.E.K.; Bos, K.K.; Stegeman, J.A.; ten Berge, A.M.; Bruining, M.; van Maarle, M.C.; Elting, M.W.; den Hollander, N.S.; Hamel, B.; et al. Genotype-Phenotype Correlations in L1 Syndrome: A Guide for Genetic Counselling and Mutation Analysis. J. Med. Genet. 2010, 47, 169–175.
- Weller, S.; Gärtner, J. Genetic and Clinical Aspects of X-Linked Hydrocephalus (L1 Disease): Mutations in the L1CAM Gene. Hum. Mutat. 2001, 18, 1–12.
- Li, S.; Wang, Y.; Sun, H.; Wang, Z.; Zhang, Q.; Wang, Y.; Yang, J.; Shi, C.; Yuan, Y.; Wang, H.; et al. Establishment of Induced Pluripotent Stem Cell Line (ZZUi033-A) of a Male with a Novel L1CAM Missense Mutation. Stem Cell Res. 2022, 59, 102663.
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. Mutations in SPG11 Are Frequent in Autosomal Recessive Spastic Paraplegia with Thin Corpus Callosum, Cognitive Decline and Lower Motor Neuron Degeneration. Brain 2008, 131, 772–784.
- Pensato, V.; Castellotti, B.; Gellera, C.; Pareyson, D.; Ciano, C.; Nanetti, L.; Salsano, E.; Piscosquito, G.; Sarto, E.; Eoli, M.; et al. Overlapping Phenotypes in Complex Spastic Paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014, 137, 1907–1920.
- Hehr, U.; Bauer, P.; Winner, B.; Schule, R.; Olmez, A.; Koehler, W.; Uyanik, G.; Engel, A.; Lenz, D.; Seibel, A.; et al. Long-term Course and Mutational Spectrum of Spatacsin-linked Spastic Paraplegia. Ann. Neurol. 2007, 62, 656–665.
- Southgate, L.; Dafou, D.; Hoyle, J.; Li, N.; Kinning, E.; Critchley, P.; Németh, A.H.; Talbot, K.; Bindu, P.S.; Sinha, S.; et al. Novel SPG11 Mutations in Asian Kindreds and Disruption of Spatacsin Function in the Zebrafish. Neurogenetics 2010, 11, 379–389.
- Crimella, C.; Arnoldi, A.; Crippa, F.; Mostacciuolo, M.L.; Boaretto, F.; Sironi, M.; D’Angelo, M.G.; Manzoni, S.; Piccinini, L.; Turconi, A.C.; et al. Point Mutations and a Large Intragenic Deletion in SPG11 in Complicated Spastic Paraplegia without Thin Corpus Callosum. J. Med. Genet. 2009, 46, 345–351.
- Chang, J.; Lee, S.; Blackstone, C. Spastic Paraplegia Proteins Spastizin and Spatacsin Mediate Autophagic Lysosome Reformation. J. Clin. Investig. 2014, 124, 5249–5262.
- Pérez-Brangulí, F.; Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Saul, D.; Rummel, C.; Dorca-Arevalo, J.; Regensburger, M.; Graef, D.; et al. Dysfunction of Spatacsin Leads to Axonal Pathology in SPG11-Linked Hereditary Spastic Paraplegia. Hum. Mol. Genet. 2014, 23, 4859–4874.
- Güner, F.; Pozner, T.; Krach, F.; Prots, I.; Loskarn, S.; Schlötzer-Schrehardt, U.; Winkler, J.; Winner, B.; Regensburger, M. Axon-Specific Mitochondrial Pathology in SPG11 Alpha Motor Neurons. Front. Neurosci. 2021, 15, 680572.
- Varga, R.-E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454.
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of Spatacsin Function Alters Lysosomal Lipid Clearance Leading to Upper and Lower Motor Neuron Degeneration. Neurobiol. Dis. 2017, 102, 21–37.
- Renvoisé, B.; Chang, J.; Singh, R.; Yonekawa, S.; FitzGibbon, E.J.; Mankodi, A.; Vanderver, A.; Schindler, A.; Toro, C.; Gahl, W.A.; et al. Lysosomal Abnormalities in Hereditary Spastic Paraplegia Types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 2014, 1, 379–389.
- Pozner, T.; Schray, A.; Regensburger, M.; Lie, D.C.; Schlötzer-Schrehardt, U.; Winkler, J.; Turan, S.; Winner, B. Tideglusib Rescues Neurite Pathology of SPG11 IPSC Derived Cortical Neurons. Front. Neurosci. 2018, 12, 914.
- Hanein, S.; Martin, E.; Boukhris, A.; Byrne, P.; Goizet, C.; Hamri, A.; Benomar, A.; Lossos, A.; Denora, P.; Fernandez, J.; et al. Identification of the SPG15 Gene, Encoding Spastizin, as a Frequent Cause of Complicated Autosomal-Recessive Spastic Paraplegia, Including Kjellin Syndrome. Am. J. Hum. Genet. 2008, 82, 992–1002.
- Goizet, C.; Boukhris, A.; Maltete, D.; Guyant-Maréchal, L.; Truchetto, J.; Mundwiller, E.; Hanein, S.; Jonveaux, P.; Roelens, F.; Loureiro, J.; et al. SPG15 Is the Second Most Common Cause of Hereditary Spastic Paraplegia with Thin Corpus Callosum. Neurology 2009, 73, 1111–1119.
- Pascual, B.; de Bot, S.T.; Daniels, M.R.; França, M.C.; Toro, C.; Riverol, M.; Hedera, P.; Bassi, M.T.; Bresolin, N.; van de Warrenburg, B.P.; et al. “Ears of the Lynx” MRI Sign Is Associated with SPG11 and SPG15 Hereditary Spastic Paraplegia. Am. J. Neuroradiol. 2019, 40, 199–203.
- Vantaggiato, C.; Crimella, C.; Airoldi, G.; Polishchuk, R.; Bonato, S.; Brighina, E.; Scarlato, M.; Musumeci, O.; Toscano, A.; Martinuzzi, A.; et al. Defective Autophagy in Spastizin Mutated Patients with Hereditary Spastic Paraparesis Type 15. Brain 2013, 136, 3119–3139.
- Słabicki, M.; Theis, M.; Krastev, D.B.; Samsonov, S.; Mundwiller, E.; Junqueira, M.; Paszkowski-Rogacz, M.; Teyra, J.; Heninger, A.-K.; Poser, I.; et al. A Genome-Scale DNA Repair RNAi Screen Identifies SPG48 as a Novel Gene Associated with Hereditary Spastic Paraplegia. PLoS Biol. 2010, 8, e1000408.
- Hirst, J.; Barlow, L.D.; Francisco, G.C.; Sahlender, D.A.; Seaman, M.N.J.; Dacks, J.B.; Robinson, M.S. The Fifth Adaptor Protein Complex. PLoS Biol. 2011, 9, e1001170.
- Hirst, J.; Edgar, J.R.; Esteves, T.; Darios, F.; Madeo, M.; Chang, J.; Roda, R.H.; Dürr, A.; Anheim, M.; Gellera, C.; et al. Loss of AP-5 Results in Accumulation of Aberrant Endolysosomes: Defining a New Type of Lysosomal Storage Disease. Hum. Mol. Genet. 2015, 24, 4984–4996.
- Ikeda, K.; Goebel, H.H.; Burck, U.; Kohlschütter, A. Ultrastructural Pathology of Human Lymphocytes in Lysosomal Disorders: A Contribution to Their Morphological Diagnosis. Eur. J. Pediatr. 1982, 138, 179–185.
- Walkley, S.U. Cellular Pathology of Lysosomal Storage Disorders. Brain Pathol. 1998, 8, 175–193.
- Parkinson-Lawrence, E.J.; Shandala, T.; Prodoehl, M.; Plew, R.; Borlace, G.N.; Brooks, D.A. Lysosomal Storage Disease: Revealing Lysosomal Function and Physiology. Physiology 2010, 25, 102–115.
- Denton, K.; Mou, Y.; Xu, C.-C.; Shah, D.; Chang, J.; Blackstone, C.; Li, X.-J. Impaired Mitochondrial Dynamics Underlie Axonal Defects in Hereditary Spastic Paraplegias. Hum. Mol. Genet. 2018, 27, 2517–2530.
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.-X.; et al. Specification of Region-Specific Neurons Including Forebrain Glutamatergic Neurons from Human Induced Pluripotent Stem Cells. PLoS ONE 2010, 5, e11853.
- Li, X.-J.; Du, Z.-W.; Zarnowska, E.D.; Pankratz, M.; Hansen, L.O.; Pearce, R.A.; Zhang, S.-C. Specification of Motoneurons from Human Embryonic Stem Cells. Nat. Biotechnol. 2005, 23, 215–221.
- Xi, J.; Liu, Y.; Liu, H.; Chen, H.; Emborg, M.E.; Zhang, S. Specification of Midbrain Dopamine Neurons from Primate Pluripotent Stem Cells. Stem Cells 2012, 30, 1655–1663.
- Murmu, R.P.; Martin, E.; Rastetter, A.; Esteves, T.; Muriel, M.-P.; El Hachimi, K.H.; Denora, P.S.; Dauphin, A.; Fernandez, J.C.; Duyckaerts, C.; et al. Cellular Distribution and Subcellular Localization of Spatacsin and Spastizin, Two Proteins Involved in Hereditary Spastic Paraplegia. Mol. Cell. Neurosci. 2011, 47, 191–202.
- Joshi, D.C.; Bakowska, J.C. Determination of Mitochondrial Membrane Potential and Reactive Oxygen Species in Live Rat Cortical Neurons. J. Vis. Exp. 2011, 51, e2704.
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003, 10, 709–717.
- Cui, M.; Tang, X.; Christian, W.V.; Yoon, Y.; Tieu, K. Perturbations in Mitochondrial Dynamics Induced by Human Mutant PINK1 Can Be Rescued by the Mitochondrial Division Inhibitor Mdivi-1. J. Biol. Chem. 2010, 285, 11740–11752.
- Hirst, J.; Bright, N.A.; Rous, B.; Robinson, M.S. Characterization of a Fourth Adaptor-Related Protein Complex. Mol. Biol. Cell 1999, 10, 2787–2802.
- Ebrahimi-Fakhari, D.; Teinert, J.; Behne, R.; Wimmer, M.; D’Amore, A.; Eberhardt, K.; Brechmann, B.; Ziegler, M.; Jensen, D.M.; Nagabhyrava, P.; et al. Defining the Clinical, Molecular and Imaging Spectrum of Adaptor Protein Complex 4-Associated Hereditary Spastic Paraplegia. Brain 2020, 143, 2929–2944.
- Hirst, J.; Irving, C.; Borner, G.H.H. Adaptor Protein Complexes AP-4 and AP-5: New Players in Endosomal Trafficking and Progressive Spastic Paraplegia. Traffic 2013, 14, 153–164.
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 Mediates Export of ATG9A from the Trans.-Golgi Network to Promote Autophagosome Formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706.
- Davies, A.K.; Itzhak, D.N.; Edgar, J.R.; Archuleta, T.L.; Hirst, J.; Jackson, L.P.; Robinson, M.S.; Borner, G.H.H. AP-4 Vesicles Contribute to Spatial Control of Autophagy via RUSC-Dependent Peripheral Delivery of ATG9A. Nat. Commun. 2018, 9, 3958.
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered Distribution of ATG9A and Accumulation of Axonal Aggregates in Neurons from a Mouse Model of AP-4 Deficiency Syndrome. PLoS Genet. 2018, 14, e1007363.
- Ivankovic, D.; Drew, J.; Lesept, F.; White, I.J.; López Doménech, G.; Tooze, S.A.; Kittler, J.T. Axonal Autophagosome Maturation Defect through Failure of ATG9A Sorting Underpins Pathology in AP-4 Deficiency Syndrome. Autophagy 2020, 16, 391–407.
- Behne, R.; Teinert, J.; Wimmer, M.; D’Amore, A.; Davies, A.K.; Scarrott, J.M.; Eberhardt, K.; Brechmann, B.; Chen, I.P.-F.; Buttermore, E.D.; et al. Adaptor Protein Complex 4 Deficiency: A Paradigm of Childhood-Onset Hereditary Spastic Paraplegia Caused by Defective Protein Trafficking. Hum. Mol. Genet. 2020, 29, 320–334.
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798.
- Davies, A.K.; Alecu, J.E.; Ziegler, M.; Vasilopoulou, C.G.; Merciai, F.; Jumo, H.; Afshar-Saber, W.; Sahin, M.; Ebrahimi-Fakhari, D.; Borner, G.H.H. AP-4-Mediated Axonal Transport Controls Endocannabinoid Production in Neurons. Nat. Commun. 2022, 13, 1058.
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.-J.; et al. Cloning of the First Sn1-DAG Lipases Points to the Spatial and Temporal Regulation of Endocannabinoid Signaling in the Brain. J. Cell Biol. 2003, 163, 463–468.
- Williams, E.-J.; Walsh, F.S.; Doherty, P. The FGF Receptor Uses the Endocannabinoid Signaling System to Couple to an Axonal Growth Response. J. Cell Biol. 2003, 160, 481–486.
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid Signaling Controls Pyramidal Cell Specification and Long-Range Axon Patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765.
- Oudin, M.J.; Hobbs, C.; Doherty, P. DAGL-dependent Endocannabinoid Signalling: Roles in Axonal Pathfinding, Synaptic Plasticity and Adult Neurogenesis. Eur. J. Neurosci. 2011, 34, 1634–1646.
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes That Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356.
- Edmison, D.; Wang, L.; Gowrishankar, S. Lysosome Function and Dysfunction in Hereditary Spastic Paraplegias. Brain Sci. 2021, 11, 152.
- Fernandopulle, M.S.; Prestil, R.; Grunseich, C.; Wang, C.; Gan, L.; Ward, M.E. Transcription Factor–Mediated Differentiation of Human IPSCs into Neurons. Curr. Protoc. Cell Biol. 2018, 79, e51.
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human IPSC-Derived Neurons. Neuron 2019, 104, 239–255.e12.
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of IPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233.
- Braulke, T.; Bonifacino, J.S. Sorting of Lysosomal Proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 605–614.
- Santangelo, S.; Bossolasco, P.; Magri, S.; Colombrita, C.; Invernizzi, S.; Gellera, C.; Nanetti, L.; Di Bella, D.; Silani, V.; Taroni, F.; et al. Generation of an IPSC Line from a Patient with Spastic Paraplegia Type 10 Carrying a Novel Mutation in KIF5A Gene. Stem Cell Res. 2023, 66, 103008.
- Wang, X.; Ma, Y.; Duan, N.; Liu, X.; Zhang, H.; Ma, J.; Liu, Y.; Sun, W.; Liu, Q. Generation of a Human Induced Pluripotent Stem Cell Line (SDUBMSi001-A) from a Hereditary Spastic Paraplegia Patient Carrying Kif1a c.773C>T Missense Mutation. Stem Cell Res. 2020, 43, 101727.
- Lin, H.-Y.; Ou-Yang, C.-H.; Lin, C.-H. Reprogramming of a Human Induced Pluripotent Stem Cell (IPSC) Line from a Patient with Neurodegeneration with Brain Iron Accumulation (NBIA) Harboring a Novel Frameshift Mutation in C19orf12 Gene. Stem Cell Res. 2020, 49, 102032.
- Leeson, H.C.; Goh, D.; Coman, D.; Wolvetang, E.J. Generation of IPSC Lines from Hereditary Spastic Paraplegia 56 (SPG56) Patients and Family Members Carrying CYP2U1 Mutations. Stem Cell Res. 2022, 64, 102917.
- Nagel, M.; Müßig, S.; Höflinger, P.; Schöls, L.; Hauser, S.; Schüle, R. Generation of the CRISPR/Cas9-Mediated KIF1C Knock-out Human IPSC Line HIHRSi003-A-1. Stem Cell Res. 2020, 49, 102059.
- Lu, Y.; Dong, E.; Yang, W.; Lai, L.; Lin, X.; Ma, L.; Chen, W.; Wang, N.; Lin, X. Generation of an Integration-Free Induced Pluripotent Stem Cell Line, FJMUi001-A, from a Hereditary Spastic Paraplegia Patient Carrying Compound Heterozygous p.P498L and p.R618W Mutations in CAPN1 (SPG76). Stem Cell Res. 2019, 34, 101354.
- Wang, L.; Brown, A. A Hereditary Spastic Paraplegia Mutation in Kinesin-1A/KIF5A Disrupts Neurofilament Transport. Mol. Neurodegener. 2010, 5, 52.
- Okada, Y.; Yamazaki, H.; Sekine-Aizawa, Y.; Hirokawa, N. The Neuron-Specific Kinesin Superfamily Protein KIF1A Is a Uniqye Monomeric Motor for Anterograde Axonal Transport of Synaptic Vesicle Precursors. Cell 1995, 81, 769–780.
- Yonekawa, Y.; Harada, A.; Okada, Y.; Funakoshi, T.; Kanai, Y.; Takei, Y.; Terada, S.; Noda, T.; Hirokawa, N. Defect in Synaptic Vesicle Precursor Transport and Neuronal Cell Death in KIF1A Motor Protein–Deficient Mice. J. Cell Biol. 1998, 141, 431–441.
- Della Vecchia, S.; Tessa, A.; Dosi, C.; Baldacci, J.; Pasquariello, R.; Antenora, A.; Astrea, G.; Bassi, M.T.; Battini, R.; Casali, C.; et al. Correction to: Monoallelic KIF1A-Related Disorders: A Multicenter Cross Sectional Study and Systematic Literature Review. J. Neurol. 2023, 270, 2345–2346.
- Tesson, C.; Nawara, M.; Salih, M.A.M.; Rossignol, R.; Zaki, M.S.; Al Balwi, M.; Schule, R.; Mignot, C.; Obre, E.; Bouhouche, A.; et al. Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2012, 91, 1051–1064.
- Pujol, C.; Legrand, A.; Parodi, L.; Thomas, P.; Mochel, F.; Saracino, D.; Coarelli, G.; Croon, M.; Popovic, M.; Valet, M.; et al. Implication of Folate Deficiency in CYP2U1 Loss of Function. J. Exp. Med. 2021, 218, e20210846.
- Husain, R.A.; Grimmel, M.; Wagner, M.; Hennings, J.C.; Marx, C.; Feichtinger, R.G.; Saadi, A.; Rostásy, K.; Radelfahr, F.; Bevot, A.; et al. Bi-Allelic HPDL Variants Cause a Neurodegenerative Disease Ranging from Neonatal Encephalopathy to Adolescent-Onset Spastic Paraplegia. Am. J. Hum. Genet. 2020, 107, 364–373.
- Ghosh, S.G.; Lee, S.; Fabunan, R.; Chai, G.; Zaki, M.S.; Abdel-Salam, G.; Sultan, T.; Ben-Omran, T.; Alvi, J.R.; McEvoy-Venneri, J.; et al. Biallelic Variants in HPDL, Encoding 4-Hydroxyphenylpyruvate Dioxygenase-like Protein, Lead to an Infantile Neurodegenerative Condition. Genet. Med. 2021, 23, 524–533.
- Wiessner, M.; Maroofian, R.; Ni, M.-Y.; Pedroni, A.; Müller, J.S.; Stucka, R.; Beetz, C.; Efthymiou, S.; Santorelli, F.M.; Alfares, A.A.; et al. Erratum to: Biallelic Variants in HPDL Cause Pure and Complicated Hereditary Spastic Paraplegia. Brain 2021, 144, e70.
- Banh, R.S.; Kim, E.S.; Spillier, Q.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Shi, G.; Jones, D.R.; Kimmelman, A.C.; Pacold, M.E. The Polar Oxy-Metabolome Reveals the 4-Hydroxymandelate CoQ10 Synthesis Pathway. Nature 2021, 597, 420–425.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
622
Revisions:
2 times
(View History)
Update Date:
12 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No