 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Brittney A Covington | -- | 2338 | 2024-03-02 23:38:08 | | | |
| 2 | Catherine Yang | + 1 word(s) | 2339 | 2024-03-04 02:01:32 | | |
Video Upload Options
Type 2 diabetes (T2D) has become a worldwide epidemic, primarily driven by obesity from overnutrition and sedentariness. Physiologically, T2D manifests as an inability of the pancreatic beta cells to produce and secrete a sufficient bolus of insulin to elicit a response in target cells to transport glucose from the blood and properly regulate glucose levels. Insulin is synthesized in the endoplasmic reticulum (ER) of pancreatic beta cells where it undergoes a series of post-translational modifications to form mature insulin. Insulin resistance requires more insulin to be produced by beta cells to compensate for these desensitized cells. Consequently, this compensation causes additional strain on beta cells. This stress primarily originates from the ER and can also trigger oxidative stress. These cellular stresses can lead to beta cell decompensation, manifested by dysfunction and eventually a loss of beta cell mass.
1. Compensation: Beta Cell Proliferation, Transdifferentiation, and Neogenesis
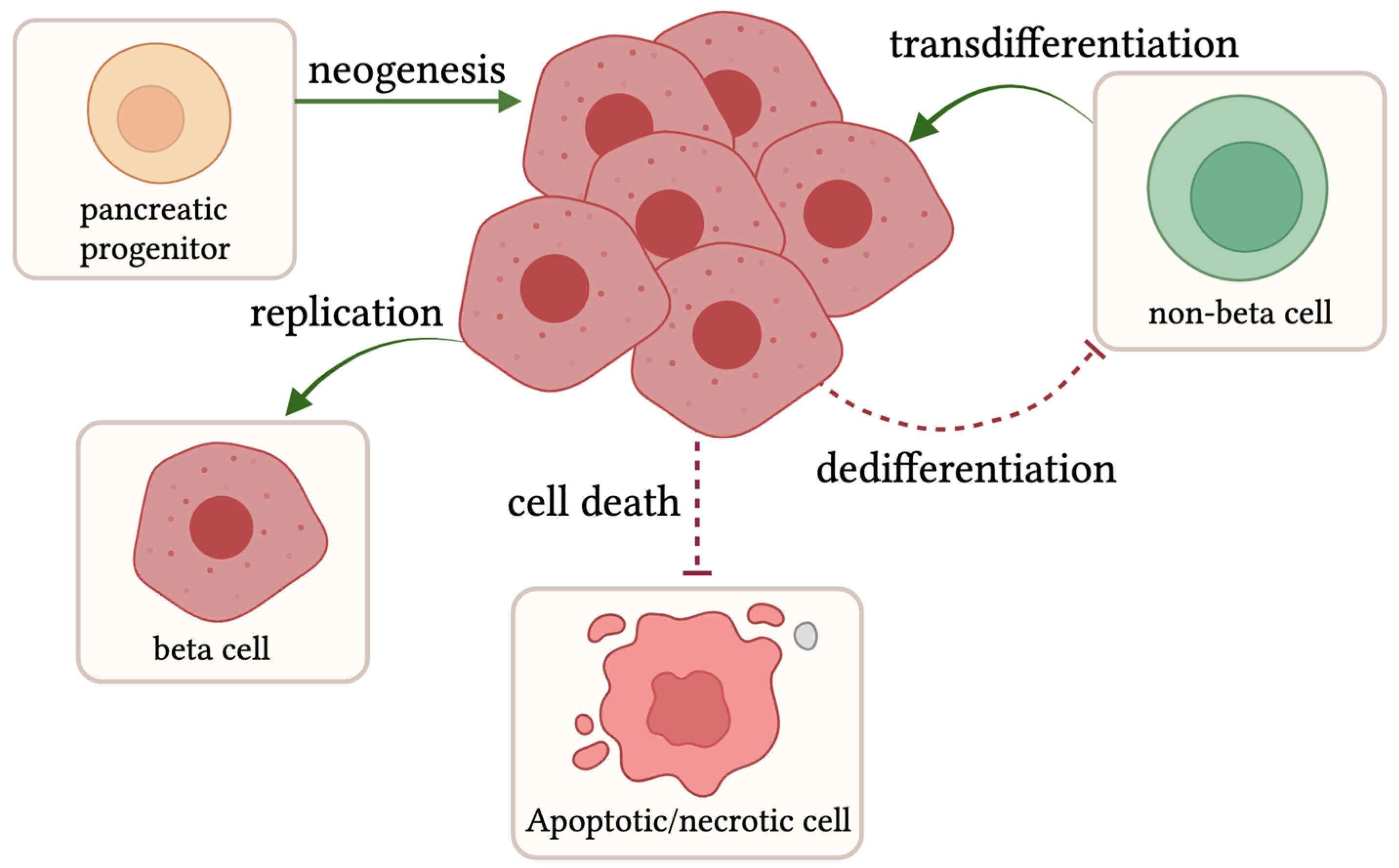
1.1. Beta Cell Expansion in Rodent Models
1.2. Human Beta Cell Expansion
1.3. Zebrafish Beta Cell Expansion
2. Decompensation: Beta Cell Death and Loss of Identity
References
- Meier, J.J. Beta cell mass in diabetes: A realistic therapeutic target? Diabetologia 2008, 51, 703–713.
- Spears, E.; Serafimidis, I.; Powers, A.C.; Gavalas, A. Debates in Pancreatic Beta Cell Biology: Proliferation versus Progenitor Differentiation and Transdifferentiation in Restoring β Cell Mass. Front. Endocrinol. 2021, 12, 722250.
- Wang, P.; Alvarez-Perez, J.C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocaña, A.; et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388.
- Yang, B.; Covington, B.A.; Chen, W. In vivo generation and regeneration of β cells in zebrafish. Cell Regen 2020, 9, 9.
- Shcheglova, E.; Blaszczyk, K.; Borowiak, M. Mitogen Synergy: An Emerging Route to Boosting Human Beta Cell Proliferation. Front. Cell Dev. Biol. 2021, 9, 734597.
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37.
- Linnemann, A.K.; Baan, M.; Davis, D.B. Pancreatic β-cell proliferation in obesity. Adv. Nutr. 2014, 5, 278–288.
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110.
- Olehnik, S.K.; Fowler, J.L.; Avramovich, G.; Hara, M. Quantitative analysis of intra- and inter-individual variability of human beta-cell mass. Sci. Rep. 2017, 7, 16398.
- Inaishi, J.; Saisho, Y. Ethnic Similarities and Differences in the Relationship between Beta Cell Mass and Diabetes. J. Clin. Med. 2017, 6, 113.
- Bonner-Weir, S.; Deery, D.; Leahy, J.L.; Weir, G.C. Compensatory Growth of Pancreatic β-Cells in Adult Rats After Short-Term Glucose Infusion. Diabetes 1989, 38, 49–53.
- Nir, T.; Melton, D.A.; Dor, Y. Recovery from diabetes in mice by β cell regeneration. J. Clin. Investig. 2007, 117, 2553–2561.
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46.
- Dalbøge, L.S.; Almholt, D.L.C.; Neerup, T.S.R.; Vassiliadis, E.; Vrang, N.; Pedersen, L.; Fosgerau, K.; Jelsing, J. Characterisation of Age-Dependent Beta Cell Dynamics in the Male db/db Mice. PLoS ONE 2013, 8, e82813.
- Pick, A.; Clark, J.; Kubstrup, C.; Levisetti, M.; Pugh, W.; Bonner-Weir, S.; Polonsky, K.S. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998, 47, 358–364.
- Kaiser, N.; Yuli, M.; Uçkaya, G.k.; Oprescu, A.I.; Berthault, M.-F.; Kargar, C.; Donath, M.Y.; Cerasi, E.; Ktorza, A. Dynamic Changes in β-Cell Mass and Pancreatic Insulin During the Evolution of Nutrition-Dependent Diabetes in Psammomys obesus: Impact of Glycemic Control. Diabetes 2005, 54, 138–145.
- Bonner-Weir, S.; Li, W.C.; Ouziel-Yahalom, L.; Guo, L.; Weir, G.C.; Sharma, A. Beta-cell growth and regeneration: Replication is only part of the story. Diabetes 2010, 59, 2340–2348.
- Wang, R.N.; Klöppel, G.; Bouwens, L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995, 38, 1405–1411.
- Bonner-Weir, S. New evidence for adult beta cell neogenesis. Cell Stem Cell 2021, 28, 1889–1890.
- Gribben, C.; Lambert, C.; Messal, H.A.; Hubber, E.L.; Rackham, C.; Evans, I.; Heimberg, H.; Jones, P.; Sancho, R.; Behrens, A. Ductal Ngn3-expressing progenitors contribute to adult β cell neogenesis in the pancreas. Cell Stem Cell 2021, 28, 2000–2008.e4.
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53, 2167–2176.
- Dai, C.; Kayton, N.S.; Shostak, A.; Poffenberger, G.; Cyphert, H.A.; Aramandla, R.; Thompson, C.; Papagiannis, I.G.; Emfinger, C.; Shiota, M.; et al. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J. Clin. Investig. 2016, 126, 1857–1870.
- Li, M.; Maddison, L.A.; Page-McCaw, P.; Chen, W. Overnutrition induces β-cell differentiation through prolonged activation of β-cells in zebrafish larvae. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E799–E807.
- Li, M.; Maddison, L.A.; Crees, Z.; Chen, W. Targeted Overexpression of CKI-Insensitive Cyclin-Dependent Kinase 4 Increases Functional β-Cell Number Through Enhanced Self-Replication in Zebrafish. Zebrafish 2013, 10, 170–176.
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449.
- Moss, J.B.; Koustubhan, P.; Greenman, M.; Parsons, M.J.; Walter, I.; Moss, L.G. Regeneration of the pancreas in adult zebrafish. Diabetes 2009, 58, 1844–1851.
- Curado, S.; Stainier, D.Y.; Anderson, R.M. Nitroreductase-mediated cell/tissue ablation in zebrafish: A spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat. Protoc. 2008, 3, 948–954.
- Pisharath, H.; Rhee, J.M.; Swanson, M.A.; Leach, S.D.; Parsons, M.J. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev. 2007, 124, 218–229.
- Ye, L.; Robertson, M.A.; Hesselson, D.; Stainier, D.Y.; Anderson, R.M. Glucagon is essential for alpha cell transdifferentiation and beta cell neogenesis. Development 2015, 142, 1407–1417.
- Delaspre, F.; Beer, R.L.; Rovira, M.; Huang, W.; Wang, G.; Gee, S.; Vitery Mdel, C.; Wheelan, S.J.; Parsons, M.J. Centroacinar Cells Are Progenitors That Contribute to Endocrine Pancreas Regeneration. Diabetes 2015, 64, 3499–3509.
- Carril Pardo, C.A.; Massoz, L.; Dupont, M.A.; Bergemann, D.; Bourdouxhe, J.; Lavergne, A.; Tarifeño-Saldivia, E.; Helker, C.S.; Stainier, D.Y.; Peers, B.; et al. A δ-cell subpopulation with a pro-β-cell identity contributes to efficient age-independent recovery in a zebrafish model of diabetes. eLife 2022, 11, e67576.
- Singh, S.P.; Chawla, P.; Hnatiuk, A.; Kamel, M.; Silva, L.D.; Spanjaard, B.; Eski, S.E.; Janjuha, S.; Olivares-Chauvet, P.; Kayisoglu, O.; et al. A single-cell atlas of de novo β-cell regeneration reveals the contribution of hybrid β/δ-cells to diabetes recovery in zebrafish. Development 2022, 149, dev199853.
- Mi, J.; Liu, K.-C.; Andersson, O. Decoding pancreatic endocrine cell differentiation and β cell regeneration in zebrafish. Sci. Adv. 2023, 9, eadf5142.
- Yu, J.; Ma, J.; Li, Y.; Zhou, Y.; Luo, L.; Yang, Y. Pax4-Ghrelin mediates the conversion of pancreatic ε-cells to β-cells after extreme β-cell loss in zebrafish. Development 2023, 150, dev201306.
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234.
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054.
- Jörns, A.; Tiedge, M.; Ziv, E.; Shafrir, E.; Lenzen, S. Gradual loss of pancreatic beta-cell insulin, glucokinase and GLUT2 glucose transporter immunoreactivities during the time course of nutritionally induced type-2 diabetes in Psammomys obesus (sand rat). Virchows Arch. 2002, 440, 63–69.
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19.
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136.
- Puff, R.; Dames, P.; Weise, M.; Göke, B.; Seissler, J.; Parhofer, K.G.; Lechner, A. Reduced proliferation and a high apoptotic frequency of pancreatic beta cells contribute to genetically-determined diabetes susceptibility of db/db BKS mice. Horm. Metab. Res. 2011, 43, 306–311.
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502.



