Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rami N. Al-Rohil | -- | 2125 | 2024-02-29 15:27:17 | | | |
| 2 | Lindsay Dong | -1 word(s) | 2124 | 2024-03-01 01:37:49 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Saade, R.; Al-Rohil, R.N. Genomic Alterations in Melanocytic Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/55742 (accessed on 29 June 2026).
Saade R, Al-Rohil RN. Genomic Alterations in Melanocytic Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/55742. Accessed June 29, 2026.
Saade, Rayan, Rami N. Al-Rohil. "Genomic Alterations in Melanocytic Tumors" Encyclopedia, https://encyclopedia.pub/entry/55742 (accessed June 29, 2026).
Saade, R., & Al-Rohil, R.N. (2024, February 29). Genomic Alterations in Melanocytic Tumors. In Encyclopedia. https://encyclopedia.pub/entry/55742
Saade, Rayan and Rami N. Al-Rohil. "Genomic Alterations in Melanocytic Tumors." Encyclopedia. Web. 29 February, 2024.
Copy Citation
Significant advances in tumor genomics have provided insight into the biology and proliferation of melanocytic tumors. Integration of clinical, histological, immunohistochemical and molecular alterations has given rise to better identification of certain melanocytic proliferations that were most likely previously lumped in the “uncertain biologic potential” category.
melanocytic tumors
melanocytic nevi
melanocytomas
genomic alterations
1. Introduction
Molecular alterations have been recognized as pivotal events driving oncogenesis in various neoplasms with biological potential spanning benign and malignant spectrums across different organs and various histogenetic categories. The field of molecular alterations in melanocytic pathology has witnessed tremendous growth in recent years. In the early phase of integrating molecular tests in melanocytic tumors, emphasis was mostly placed on BRAF mutations in metastatic melanoma to help provide targeted therapeutic agents. In recent years, the field has expanded drastically in recognizing molecular alterations and fusions corresponding to certain histopathologic features and certain melanocytic subtypes.
2. Spitz Tumors
2.1. HRAS-Driven Spitz Tumors
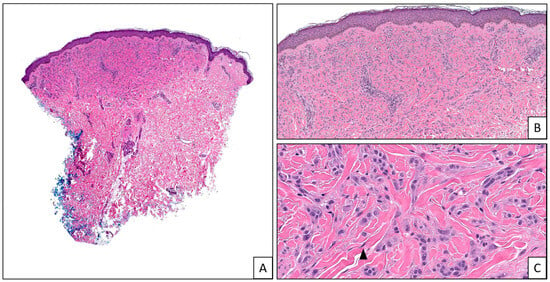
Genomic studies have found that true Spitz lineage, unlike conventional nevi and melanomas, lack BRAF- or NRAS-activating mutations and instead often harbor kinase fusions and characteristic HRAS mutations and/or amplifications, which have become defining diagnostic criteria in the 2018 WHO classification of skin tumors [1]. HRAS is a proto-oncogene located on chromosome 11p that is part of to the RAS family of oncogenes and encodes a GTPase, which is a member of the small GTPase family that, upon activation by growth factors, stimulates multiple downstream pathways, namely MAP kinase and PI3K-AKT signaling, to promote cell proliferation and survival [2][3]. HRAS mutations with or without 11p amplification define a subtype of Spitz neoplasms with distinctive clinical and morphologic features [4]. Clinically, these lesions tend to be symmetric and raised with a predilection to the head and neck, and extremities [5]. Activating mutations in HRAS have also been described in association with grouped patterns of Spitz nevi, especially agminated Spitz nevi, whether arising in a nevus spilus or not [6][7]. Histologically, HRAS-mutated Spitz nevi are variably cellular and characteristically show an intradermal component with desmoplastic stroma (Figure 1), particularly when 11p amplification is present [8][9].
Figure 1. Desmoplastic Spitz nevus: (A) a dermal-based melanocytic proliferation (2×). (B). The melanocytes induce background dermal fibrosis/desmoplasia (4×). (C). The melanocytes display a Spitzoid morphology (arrow head) with uniform atypia and no significant pleomorphism or mitotic activity (20×). These nevi are usually driven by HRAS mutations.
2.2. ALK-Fused Spitz Tumors
Tyrosine kinase fusion-associated Spitz neoplasms are an emerging category of tumors with specific phenotypic patterns that can improve the accuracy of identifying a tumor as belonging to the Spitz family. These fusions constitute primary driver events that seem to correlate with specific histologic features of each melanocytic neoplasm.
The anaplastic lymphoma kinase (ALK) gene is an oncogene located on chromosome 2p and synthesizes a tyrosine kinase receptor that belongs to the insulin receptor superfamily [10]. Activating genomic alterations in this gene, including fusions with several partner genes, promote cell proliferation by activating MAPK, PI3K, and JAK3 signaling pathways [11][12].
Clinically, ALK-fused Spitz neoplasms characteristically present as solitary, large, amelanotic, polypoid lesions on the extremities of young patients [13]. Morphologically, these tumors are commonly dome/wedge-shaped compound lesions with a predominant dermal melanocytic component that can show an infiltrative pattern [14][15][16]. Cells are typically non-pigmented, spindled with pericellular clefts or mixed with an epithelioid component and frequently show vesicular nuclei with prominent nucleoli and an amphophilic cytoplasm [14][15][16][17].
2.3. ROS1-Fused Spitz Tumors
ROS1 is a proto-oncogene found on chromosome 6q and synthesizes an orphan receptor tyrosine kinase that may activate multiple pathways involved in cell survival and transformation, namely RAS-RAF, JAK3-STAT and PI3K-AKT-mTOR pathwaysv [18]. Ros1 fusion proteins resulting from the fusion of ROS1 gene with several partner genes frequently lead to constitutive activation of Ros1 signaling and are a relatively common occurrence in Spitz melanocytic tumors [19].
Clinically, ROS1-fused Spitz neoplasms are present in young adults as dome-shaped erythematous papules that show a predilection to the lower extremities but can be found all throughout the body [20]. Morphological features are rather non-specific [20]; however, most neoplasms described in the literature show a compound plaque-like to nodular architecture with conspicuous junctional melanocytic nests, trans-epidermal elimination, and occasional involvement of the adnexal epithelium (including acrosyringial units) [17][20][21] .
2.4. NTRK-Fused Spitz Tumors
The neurotrophic receptor tyrosine kinase (NTRK) genes NTRK1, NTRK2, and NTRK3 are oncogenes that reside on chromosomes 1q, 9q, and 15q, respectively, and encode single-pass receptor tyrosine kinase proteins that belong to the TRK family of tyrosine kinase receptors [22]. Upon binding to neurotrophins, these cell surface receptors can initiate signaling cascades through various mechanisms, including MAPK, PI3K-AKT, and PLCγ1 pathways, leading to cell growth and differentiation [22][23]. Most oncogenic fusions involving the NTRK gene family result in chimeric proteins with a retained kinase domain and an acquired dimerizing domain, leading to ligand-independent activation of downstream pathways [24]. NTRK1, NTRK2, and NTRK3 fusions have all been reported in Spitz neoplasms, with NTRK1 being the most common among them [14][17][24][25][26][27][28][29][30].
Spitz tumors with NTRK1 fusions commonly show a symmetric, compound or intradermal, flat or wedge-shaped architecture and are composed of small spindled to epithelioid melanocytes arranged in lobulated junctional and dermal nests that are organized in a back-to-back pattern forming larger nests [14][25][26]. Characteristically, these lesions show elongated, thin/filigree-like rete ridges, frequent Kamino bodies, rosette-like structures, and extreme maturation and are often associated with a lymphocytic infiltrate [14][16][25][26].
NTRK2-fused Spitz neoplasms are rare. In one case report of an NTRK2-fused Spitz nevus, the morphologic features were those of a pigmented spindle cell nevus with essentially large junctional nests of spindled pigmented melanocytes with an abundant eosinophilic cytoplasm, no nuclear atypia and associated hyperplastic epidermis and Kamino bodies [28].
2.5. MET-Fused Spitz Tumors
MET is a proto-oncogene found on chromosome 7q that synthesizes a receptor tyrosine kinase that activates MAPK, PI3K-AKT, PLCγ1, β-catenin, and STAT pathways to promote cell proliferation and motility [31]. The number of cases of MET-fused Spitz neoplasms in the literature is limited; however, all the cases reported thus far harbored a breakpoint in intron 14 of the MET gene, which contains the regulatory domain of the Met protein and is located upstream of the kinase domain, which is preserved in the fusion protein [32].
2.6. RET-Fused Spitz Tumors
RET is a proto-oncogene that resides on chromosome 10q and encodes a receptor tyrosine kinase that can activate MAPK, PI3K-AKT, and PLCγ1 signaling pathways, thereby regulating cell growth and differentiation [33][34]. RET fusions with several partner genes have been reported in a minor subset of Spitz melanocytic neoplasms [19][35]. Spitz neoplasms with RET fusions are not currently associated with specific morphological features. The reported cases are often well-circumscribed, symmetric, plaque-like compound proliferations containing large expansile discohesive nests of small to intermediate-sized, monotonous, epithelioid melanocytes with mild to moderate cytologic atypia [19][35].
2.7. BRAF-Fused Spitz Tumors
Spitz neoplasms with serine/threonine kinase fusions or mutations are a subtype of tumors characterized by worrisome histologic features, higher grade cytologic atypia and a greater likelihood of being classified as atypical Spitz tumor (AST) or malignant Spitz tumor (MST) [14][25][32][36][37][38][39][40][41].
BRAF is a proto-oncogene located on chromosome 7q that encodes Braf protein, a member of the Raf family of serine/threonine protein kinases, which signals through the MAK kinase pathway to regulate cell proliferation and cell growth [42][43].
Clinically, Spitz neoplasms with BRAF fusions most commonly present in young females as pink papules on the extremities [37]. Morphologically, these lesions can be compound or intradermal with variable plaque-like, wedge-shaped, or nodular configurations and are mostly composed of large epithelioid melanocytes with an abundant amphophilic cytoplasm, vesicular pleomorphic nuclei and prominent nucleoli [14][25][36][37][38][39]. Cytologic atypia is frequently moderate to severe, and high mitotic activity is usually evident [14][25][36][37][38][39].
2.8. MAP3K8-Mutated Spitz Tumors
The mitogen-activated protein kinase kinase kinase 8 (MAP3K8) gene is located on chromosome 10p. It encodes a serine/threonine and tyrosine kinase that is primarily expressed by the immune system and is activated by TNF-alpha, IL1R, and toll-like receptors to promote signaling through activation of the RAF-MEK1/2-ERK1/2 pathway [44][45][46].
Clinically, MAP3K8-fused Spitz neoplasms usually present as pigmented exophytic lesions on the lower extremities of patients of all ages, with a slight female predilection [47][48]. Morphologically, most of these lesions are nodular or dome-shaped, asymmetric with overlying epidermal hyperplasia, and show a compound melanocytic proliferation with a predominantly nested junctional component [17][25][47][48][49][50][51]. Melanocytes are invariably epithelioid with large, uniform nuclei, prominent nucleoli and an abundant eosinophilic cytoplasm [17][47][51].
2.9. MAP2K1 Mutated Spitz Tumors
Mitogen-activated protein kinase kinase 1 (MAP2K1) is a proto-oncogene that resides on chromosome 15q. It encodes MEK1, a serine-threonine kinase downstream of RAF in the RAS-RAF-MEK-ERK pathway, which in turn activates the MAPK pathway in cell proliferation and differentiation [50][52]. Although rare, MAP2K1 mutations, particularly in-frame deletions in exons 2 and 3, have been reported in Spitz neoplasms [53]. These deletions frequently lead to the inactivation of the autoinhibitory domain of the MEK1 protein resulting in unopposed activation of the kinase domain [53].
Clinically, MAP2K1-mutated Spitz neoplasms are mostly present on the lower extremities of young females as small, pigmented, flat, or mildly elevated lesions [50][53][54][55]. No distinctive histomorphological features have been established for these lesions given the small number of cases. However, recurring histologic characteristics among the cases described include a wedge-shaped compound or intradermal melanocytic proliferation composed of large epithelioid cells with vesicular nuclei and moderate to severe nuclear pleomorphism arranged in nests and showing a plexiform growth pattern, poor maturation and a tendency to converge around the adnexal structures and neurovascular bundles [50][53][54][55].
3. Blue Nevi
Blue nevi are associated with activating mutations in the Gαq pathway, namely point mutations in GNAQ or GNA11 and less commonly hotspot mutations in CYSLTR2 or fusions of protein kinase C (PKC) isoforms [56][57][58][59]. GNAQ and GNA11 are oncogenes located on chromosomes 9q and 19p, respectively, and encode G protein subunits alpha q and alpha 11 consecutively, which are guanine-binding proteins (G proteins) that are activated upon ligand binding to G-protein-associated receptors and function in downstream signaling [60]. GNAQ and GNA11 hotspot mutations alter intrinsic GTPase activity, leading to constitutive pathway activation [60][61][62].
Clinically, blue nevi typically present as grayish blue-black macules, papules, or nodules on the head, buttock, or lower extremities and are more frequent in young adult females. The blue color is caused by the Tyndall effect, where light preferentially scatters shorter wavelengths by the melanin in the dermis [63][64][65]. Histologically, multiple variants are present; however, in all cases, these tumors are almost always intradermal and are characterized by bipolar spindle dendritic pigmented melanocytes and melanophages, often growing in between sclerotic collagen bundles [66]. The cellular variant is usually biphasic and contains distinct cellular areas of plump, spindled to oval melanocytes with clear or finely pigmented melanocytes arranged in fascicles and nests [66][67].
4. Deep Penetrating Melanocytoma (DPM)
Deep penetrating melanocytomas (DPMs) are of the low cumulative sun damage (CSD) pathways that, in addition to activating mutations in BRAF or MAP2K1, harbor mutations that result in constitutive activation of the Wnt/β-catenin pathway, most often point mutations in catenin beta 1 (CTNNB1) gene on chromosome 3p, a component of the cadherin-based adherens junction, which prevent its degradation, but alternatively biallelic inactivation of adenomatous polyposis coli (APC) gene on chromosome 5q, a major component of the CTNNB1 degradation complex [68][69][70]. CTNNB1 imbalance is implicated in tumor growth, progression, and survival advantage [71].
Clinically, DPNs present in young to middle-aged patients as pigmented papules or nodules, usually on the face, neck, or shoulder [72][73][74]. Histologically, these lesions can arise in a pre-existing compound nevus. They are characterized by a wedge-shaped silhouette, an inconspicuous junctional component, and a cellular dermal component that can extend to the reticular dermis or even subcutis [72][73][74][75]. Melanocytes are commonly arranged in fascicles or nests and typically show heavy pigmentation and a plump epithelioid to spindle cell morphology with a lack of maturation [72][73][74][75]. Mild cytologic atypia, nuclear pleomorphism, and occasional dermal mitotic figures are not uncommon [72][73][74][75].
5. Pigmented Epithelioid Melanocytoma (PEM)
Pigmented epithelioid melanocytomas (PEM) are intermediate-grade melanocytic tumors of the low-CSD pathway that can harbor either biallelic inactivation of the protein kinase cAMP-dependent type I regulatory subunit alpha (PRKAR1A) gene on chromosome 17q, a major component of protein kinase A (PKA), which mediates cAMP-dependent signaling and regulates PKA activation, or fusions in the protein kinase C alpha (PRKCA) gene, a member of the protein kinase C (PKC) family of serine/threonine kinases, which is involved in a number of essential cellular processes including proliferation, differentiation, survival, and migration [76][77][78][79][80][81][82][83][84].
Clinically, PEM is classically present as pigmented, blue to blue-black, dome-shaped, papular or nodular lesions, mostly on the extremities, head and neck, and trunk of young adults, children, and infants [80][81][85]. Histologically, these lesions typically show a nodular or wedge-shaped proliferation of heavily pigmented large multinucleated and small epithelioid, spindled, and dendritic melanocytes and melanophages with the majority having overlying epidermal hyperplasia [76][78][80][81][85]. The junctional component is usually inconspicuous, and the dermal component characteristically consists of melanocytes with large vesicular nuclei and prominent nucleoli arranged in single cells and small nests that show a lack of maturation [76][78][80][81][85].
References
- Barnhill, R.; Bahrami, A.; Bastian, B.C.; Busam, K.J.; Cerroni, L.; de la Fouchardiere, A.; Elder, D.E.; Gerami, P.; Lazova, R.; Schmidt, B.; et al. 2.-8.A. Malignant Spitz tumour (Spitz melanoma). In WHO Classification of Skin Tumours; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; World Health Organization: Geneva, Switzerland, 2018; Volume 4, pp. 108–110.
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes. Cancer 2011, 2, 344–358.
- Shu, L.; Wang, D.; Saba, N.F.; Chen, Z.G. A Historic Perspective and Overview of H-Ras Structure, Oncogenicity, and Targeting. Mol. Cancer Ther. 2020, 19, 999–1007.
- van Dijk, M.C.; Bernsen, M.R.; Ruiter, D.J. Analysis of mutations in B-RAF, N-RAS, and H-RAS genes in the differential diagnosis of Spitz nevus and spitzoid melanoma. Am. J. Surg. Pathol. 2005, 29, 1145–1151.
- Lezcano, C.M.; Yeh, I.; Eslamdoost, N.; Fang, Y.; LeBoit, P.E.; McCalmont, T.H.; Moy, A.P.; Zhang, Y.; Busam, K.J. Expanding the Spectrum of Microscopic and Cytogenetic Findings Associated With Spitz Tumors With 11p Gains. Am. J. Surg. Pathol. 2021, 45, 277–285.
- Porubsky, C.; Teer, J.K.; Zhang, Y.; Deschaine, M.; Sondak, V.K.; Messina, J.L. Genomic analysis of a case of agminated Spitz nevi and congenital-pattern nevi arising in extensive nevus spilus. J. Cutan. Pathol. 2018, 45, 180–183.
- Sarin, K.Y.; Sun, B.K.; Bangs, C.D.; Cherry, A.; Swetter, S.M.; Kim, J.; Khavari, P.A. Activating HRAS mutation in agminated Spitz nevi arising in a nevus spilus. JAMA Dermatol. 2013, 149, 1077–1081.
- van Engen-van Grunsven, A.C.; van Dijk, M.C.; Ruiter, D.J.; Klaasen, A.; Mooi, W.J.; Blokx, W.A. HRAS-mutated Spitz tumors: A subtype of Spitz tumors with distinct features. Am. J. Surg. Pathol. 2010, 34, 1436–1441.
- Bastian, B.C.; LeBoit, P.E.; Pinkel, D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am. J. Pathol. 2000, 157, 967–972.
- Barreca, A.; Lasorsa, E.; Riera, L.; Machiorlatti, R.; Piva, R.; Ponzoni, M.; Kwee, I.; Bertoni, F.; Piccaluga, P.P.; Pileri, S.A.; et al. European T-Cell Lymphoma Study Group. Anaplastic lymphoma kinase in human cancer. J. Mol. Endocrinol. 2011, 47, R11–R23.
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27 (Suppl. 3), iii4–iii15.
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047.
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591.
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzì, A.E.; Gerami, P. A Comparison of Morphologic and Molecular Features of BRAF, ALK, and NTRK1 Fusion Spitzoid Neoplasms. Am. J. Surg. Pathol. 2017, 41, 491–498.
- Busam, K.J.; Kutzner, H.; Cerroni, L.; Wiesner, T. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2014, 38, 925–933.
- Kiuru, M.; Jungbluth, A.; Kutzner, H.; Wiesner, T.; Busam, K.J. Spitz Tumors: Comparison of Histological Features in Relationship to Immunohistochemical Staining for ALK and NTRK1. Int. J. Surg. Pathol. 2016, 24, 200–206.
- Kervarrec, T.; Pissaloux, D.; Tirode, F.; Samimi, M.; Jacquemus, J.; Castillo, C.; de la Fouchardière, A. Morphologic features in a series of 352 Spitz melanocytic proliferations help predict their oncogenic drivers. Virchows Arch. 2022, 480, 369–382.
- Davies, K.D.; Doebele, R.C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 2013, 19, 4040–4045.
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116.
- Gerami, P.; Kim, D.; Compres, E.V.; Zhang, B.; Khan, A.U.; Sunshine, J.C.; Quan, V.L.; Busam, K. Clinical, morphologic, and genomic findings in ROS1 fusion Spitz neoplasms. Mod. Pathol. 2021, 34, 348–357.
- Donati, M.; Kastnerova, L.; Martinek, P.; Grossmann, P.; Sticová, E.; Hadravský, L.; Torday, T.; Kyclova, J.; Michal, M.; Kazakov, D.V. Spitz Tumors with ROS1 Fusions: A Clinicopathological Study of 6 Cases, Including FISH for Chromosomal Copy Number Alterations and Mutation Analysis Using Next-Generation Sequencing. Am. J. Dermatopathol. 2020, 42, 92–102.
- Rubin, J.B.; Segal, R.A. Growth, survival and migration: The Trk to cancer. Cancer Treat. Res. 2003, 115, 1–18.
- Wiesner, T.; Kutzner, H.; Cerroni, L.; Mihm MCJr Busam, K.J.; Murali, R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology. 2016, 48, 113–131.
- Wang, L.; Busam, K.J.; Benayed, R.; Cimera, R.; Wang, J.; Denley, R.; Rao, M.; Aryeequaye, R.; Mullaney, K.; Cao, L.; et al. Identification of NTRK3 Fusions in Childhood Melanocytic Neoplasms. J. Mol. Diagn. 2017, 19, 387–396.
- Raghavan, S.S.; Peternel, S.; Mully, T.W.; North, J.P.; Pincus, L.B.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod. Pathol. 2020, 33, 1122–1134.
- Yeh, I.; Busam, K.J.; McCalmont, T.H.; LeBoit, P.E.; Pissaloux, D.; Alberti, L.; de la Fouchardière, A.; Bastian, B.C. Filigree-like Rete Ridges, Lobulated Nests, Rosette-like Structures, and Exaggerated Maturation Characterize Spitz Tumors With NTRK1 Fusion. Am. J. Surg. Pathol. 2019, 43, 737–746.
- de la Fouchardière, A.; Tee, M.K.; Peternel, S.; Valdebran, M.; Pissaloux, D.; Tirode, F.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; et al. Fusion partners of NTRK3 affect subcellular localization of the fusion kinase and cytomorphology of melanocytes. Mod. Pathol. 2021, 34, 735–747.
- Goto, K.; Pissaloux, D.; Tirode, F.; de la Fouchardière, A. Spitz nevus with a novel TFG-NTRK2 fusion: The first case report of NTRK2-rearranged Spitz/Reed nevus. J. Cutan. Pathol. 2021, 48, 1193–1196.
- Yeh, I.; Tee, M.K.; Botton, T.; Shain, A.H.; Sparatta, A.J.; Gagnon, A.; Vemula, S.S.; Garrido, M.C.; Nakamaru, K.; Isoyama, T.; et al. NTRK3 kinase fusions in Spitz tumours. J. Pathol. 2016, 240, 282–290.
- Cappellesso, R.; Nozzoli, F.; Zito Marino, F.; Simi, S.; Castiglione, F.; De Giorgi, V.; Cota, C.; Senetta, R.; Scognamiglio, G.; Anniciello, A.M.; et al. NTRK Gene Fusion Detection in Atypical Spitz Tumors. Int. J. Mol. Sci. 2021, 22, 12332.
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3 (Suppl. 1), S21–S35.
- Lee, C.Y.; Sholl, L.M.; Zhang, B.; Merkel, E.A.; Amin, S.M.; Guitart, J.; Gerami, P. Atypical Spitzoid Neoplasms in Childhood: A Molecular and Outcome Study. Am. J. Dermatopathol. 2017, 39, 181–186.
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186.
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 151–167.
- Kim, D.; Compres, E.V.; Zhang, B.; Khan, A.U.; Sunshine, J.C.; Quan, V.L.; Gerami, P. A Series of RET Fusion Spitz Neoplasms with Plaque-Like Silhouette and Dyscohesive Nesting of Epithelioid Melanocytes. Am. J. Dermatopathol. 2021, 43, 243–251.
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345.
- Kim, D.; Khan, A.U.; Compres, E.V.; Zhang, B.; Sunshine, J.C.; Quan, V.L.; Gerami, P. BRAF fusion Spitz neoplasms; clinical morphological, and genomic findings in six cases. J. Cutan. Pathol. 2020, 47, 1132–1142.
- Perron, E.; Pissaloux, D.; Neub, A.; Hohl, D.; Tartar, M.D.; Mortier, L.; Alberti, L.; de la Fouchardiere, A. Unclassified sclerosing malignant melanomas with AKAP9-BRAF gene fusion: A report of two cases and review of BRAF fusions in melanocytic tumors. Virchows Arch. 2018, 472, 469–476.
- Donati, M.; Kastnerova, L.; Ptakova, N.; Michal, M.; Kazakov, D.V. Polypoid Atypical Spitz Tumor with a Fibrosclerotic Stroma, CLIP2-BRAF Fusion, and Homozygous Loss of 9p21. Am. J. Dermatopathol. 2020, 42, 204–207.
- Wu, G.; Barnhill, R.L.; Lee, S.; Li, Y.; Shao, Y.; Easton, J.; Dalton, J.; Zhang, J.; Pappo, A.; Bahrami, A. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod. Pathol. 2016, 29, 359–369.
- Zarabi, S.K.; Azzato, E.M.; Tu, Z.J.; Ni, Y.; Billings, S.D.; Arbesman, J.; Funchain, P.; Gastman, B.; Farkas, D.H.; Ko, J.S. Targeted next generation sequencing (NGS) to classify melanocytic neoplasms. J. Cutan. Pathol. 2020, 47, 691–704.
- Jarkowski A 3rd Khushalani, N.I. BRAF and beyond: Tailoring strategies for the individual melanoma patient. J. Carcinog. 2014, 13, 1.
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199.
- Xu, D.; Matsumoto, M.L.; McKenzie, B.S.; Zarrin, A.A. TPL2 kinase action and control of inflammation. Pharmacol. Res. 2018, 129, 188–193.
- Salmeron, A.; Ahmad, T.B.; Carlile, G.W.; Pappin, D.; Narsimhan, R.P.; Ley, S.C. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 1996, 15, 817–826.
- Hagemann, D.; Troppmair, J.; Rapp, U.R. Cot protooncoprotein activates the dual specificity kinases MEK-1 and SEK-1 and induces differentiation of PC12 cells. Oncogene 1999, 18, 1391–1400.
- Houlier, A.; Pissaloux, D.; Masse, I.; Tirode, F.; Karanian, M.; Pincus, L.B.; McCalmont, T.H.; LeBoit, P.E.; Bastian, B.C.; Yeh, I.; et al. Melanocytic tumors with MAP3K8 fusions: Report of 33 cases with morphological-genetic correlations. Mod. Pathol. 2020, 33, 846–857.
- Newman, S.; Fan, L.; Pribnow, A.; Silkov, A.; Rice, S.V.; Lee, S.; Shao, Y.; Shaner, B.; Mulder, H.; Nakitandwe, J.; et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat. Med. 2019, 25, 597–602.
- Quan, V.L.; Zhang, B.; Zhang, Y.; Mohan, L.S.; Shi, K.; Wagner, A.; Kruse, L.; Taxter, T.; Beaubier, N.; White, K.; et al. Integrating Next-Generation Sequencing with Morphology Improves Prognostic and Biologic Classification of Spitz Neoplasms. J. Investig. Dermatol. 2020, 140, 1599–1608.
- Quan, V.L.; Zhang, B.; Mohan, L.S.; Shi, K.; Isales, M.C.; Panah, E.; Taxter, T.J.; Beaubier, N.; White, K.; Gerami, P. Activating Structural Alterations in MAPK Genes Are Distinct Genetic Drivers in a Unique Subgroup of Spitzoid Neoplasms. Am. J. Surg. Pathol. 2019, 43, 538–548.
- Newman, S.; Pappo, A.; Raimondi, S.; Zhang, J.; Barnhill, R.; Bahrami, A. Pathologic Characteristics of Spitz Melanoma With MAP3K8 Fusion or Truncation in a Pediatric Cohort. Am. J. Surg. Pathol. 2019, 43, 1631–1637.
- Bromberg-White, J.L.; Andersen, N.J.; Duesbery, N.S. MEK genomics in development and disease. Brief. Funct. Genom. 2012, 11, 300–310.
- Sunshine, J.C.; Kim, D.; Zhang, B.; Compres, E.V.; Khan, A.U.; Busam, K.J.; Gerami, P. Melanocytic Neoplasms with MAP2K1 in Frame Deletions and Spitz Morphology. Am. J. Dermatopathol. 2020, 42, 923–931.
- Donati, M.; Nosek, D.; Waldenbäck, P.; Martinek, P.; Jonsson, B.A.; Galgonkova, P.; Hawawrehova, M.; Berouskova, P.; Kastnerova, L.; Persichetti, P.; et al. MAP2K1-Mutated Melanocytic Neoplasms With a SPARK-Like Morphology. Am. J. Dermatopathol. 2021, 43, 412–417.
- Kerckhoffs, K.G.P.; Aallali, T.; Ambarus, C.A.; Sigurdsson, V.; Jansen, A.M.L.; Blokx, W.A.M. Expanding spectrum of “spitzoid” lesions: A small series of 4 cases with MAP2K1 mutations. Virchows Arch. 2021, 479, 195–202.
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602.
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199.
- Möller, I.; Murali, R.; Müller, H.; Wiesner, T.; Jackett, L.A.; Scholz, S.L.; Cosgarea, I.; van de Nes, J.A.; Sucker, A.; Hillen, U.; et al. Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod. Pathol. 2017, 30, 350–356.
- Goto, K.; Pissaloux, D.; Paindavoine, S.; Tirode, F.; de la Fouchardière, A. CYSLTR2-mutant Cutaneous Melanocytic Neoplasms Frequently Simulate “Pigmented Epithelioid Melanocytoma”, Expanding the Morphologic Spectrum of Blue Tumors: A Clinicopathologic Study of 7 Cases. Am. J. Surg. Pathol. 2019, 43, 1368–1376.
- Urtatiz, O.; Van Raamsdonk, C.D. Gnaq and Gna11 in the Endothelin Signaling Pathway and Melanoma. Front. Genet. 2016, 7, 59.
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424.
- Livingstone, E.; Zaremba, A.; Horn, S.; Ugurel, S.; Casalini, B.; Schlaak, M.; Hassel, J.C.; Herbst, R.; Utikal, J.S.; Weide, B.; et al. GNAQ and GNA11 mutant nonuveal melanoma: A subtype distinct from both cutaneous and uveal melanoma. Br. J. Dermatol. 2020, 183, 928–939.
- Zembowicz, A.; Mihm, M.C. Dermal dendritic melanocytic proliferations: An update. Histopathology 2004, 45, 433–451.
- Argenziano, G.; Zalaudek, I.; Ferrara, G.; Hofmann-Wellenhof, R.; Soyer, H.P. Proposal of a new classification system for melanocytic naevi. Br. J. Dermatol. 2007, 157, 217–227.
- Temple-Camp, C.R.; Saxe, N.; King, H. Benign and malignant cellular blue nevus. A clinicopathological study of 30 cases. Am. J. Dermatopathol. 1988, 10, 289–296.
- Murali, R.; McCarthy, S.W.; Scolyer, R.A. Blue nevi and related lesions: A review highlighting atypical and newly described variants, distinguishing features and diagnostic pitfalls. Adv. Anat. Pathol. 2009, 16, 365–382.
- Rodriguez, H.A.; Ackerman, L.V. Cellular blue nevus: Clinicopathologic study of forty-five cases. Cancer 1968, 21, 393–405.
- Yeh, I.; Lang, U.E.; Durieux, E.; Tee, M.K.; Jorapur, A.; Shain, A.H.; Haddad, V.; Pissaloux, D.; Chen, X.; Cerroni, L.; et al. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat. Commun. 2017, 8, 644.
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14.
- Akiyama, T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev. 2000, 11, 273–282.
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736.
- Seab, J.A., Jr.; Graham, J.H.; Helwig, E.B. Deep penetrating nevus. Am. J. Surg. Pathol. 1989, 13, 39–44.
- Robson, A.; Morley-Quante, M.; Hempel, H.; McKee, P.H.; Calonje, E. Deep penetrating naevus: Clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology 2003, 43, 529–537.
- Luzar, B.; Calonje, E. Deep penetrating nevus: A review. Arch. Pathol. Lab. Med. 2011, 135, 321–326.
- Mehregan, D.A.; Mehregan, A.H. Deep penetrating nevus. Arch. Dermatol. 1993, 129, 328–331.
- Zembowicz, A.; Carney, J.A.; Mihm, M.C. Pigmented epithelioid melanocytoma: A low-grade melanocytic tumor with metastatic potential indistinguishable from animal-type melanoma and epithelioid blue nevus. Am. J. Surg. Pathol. 2004, 28, 31–40.
- Cohen, J.N.; Joseph, N.M.; North, J.P.; Onodera, C.; Zembowicz, A.; LeBoit, P.E. Genomic Analysis of Pigmented Epithelioid Melanocytomas Reveals Recurrent Alterations in PRKAR1A, and PRKCA Genes. Am. J. Surg. Pathol. 2017, 41, 1333–1346.
- Bahrami, A.; Lee, S.; Wu, G.; Kerstetter, J.; Rahvar, M.; Li, X.; Easton, J.; Zhang, J.; Barnhill, R.L. Pigment-Synthesizing Melanocytic Neoplasm With Protein Kinase C Alpha (PRKCA) Fusion. JAMA Dermatol. 2016, 152, 318–322.
- Isales, M.C.; Mohan, L.S.; Quan, V.L.; Garfield, E.M.; Zhang, B.; Shi, K.; Arva, N.; Beaubier, N.; Yazdan, P.; White, K.; et al. Distinct Genomic Patterns in Pigmented Epithelioid Melanocytoma: A Molecular and Histologic Analysis of 16 Cases. Am. J. Surg. Pathol. 2019, 43, 480–488.
- Cohen, J.N.; Yeh, I.; Mully, T.W.; LeBoit, P.E.; McCalmont, T.H. Genomic and Clinicopathologic Characteristics of PRKAR1A-inactivated Melanomas: Toward Genetic Distinctions of Animal-type Melanoma/Pigment Synthesizing Melanoma. Am. J. Surg. Pathol. 2020, 44, 805–816.
- Benton, S.; Zhao, J.; Asadbeigi, S.; Kim, D.; Zhang, B.; Gerami, P. Pigmented Epithelioid Melanocytoma: Morphology and Molecular Drivers. Surg. Pathol. Clin. 2021, 14, 285–292.
- Berthon, A.S.; Szarek, E.; Stratakis, C.A. PRKACA: The catalytic subunit of protein kinase A and adrenocortical tumors. Front. Cell Dev. Biol. 2015, 3, 26.
- Michie, A.M.; Nakagawa, R. The link between PKCalpha regulation and cellular transformation. Immunol. Lett. 2005, 96, 155–162.
- Nakashima, S. Protein kinase C alpha (PKC alpha): Regulation and biological function. J. Biochem. 2002, 132, 669–675.
- Williams, E.A.; Shah, N.; Danziger, N.; Montesion, M.; Sokol, E.S.; Pavlick, D.C.; Miller, V.A.; Ross, J.S.; Elvin, J.A.; Tse, J.Y. Clinical, histopathologic, and molecular profiles of PRKAR1A-inactivated melanocytic neoplasms. J. Am. Acad. Dermatol. 2021, 84, 1069–1071.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
535
Revisions:
2 times
(View History)
Update Date:
01 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No