 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manolis Mavroidis | -- | 2488 | 2024-02-29 08:29:54 | | | |
| 2 | Lindsay Dong | Meta information modification | 2488 | 2024-02-29 09:02:18 | | |
Video Upload Options
Autosomal Dominant Polycystic Kidney Disease (ADPKD) stands as the most prevalent hereditary renal disorder in humans, ultimately culminating in end-stage kidney disease. Animal models carrying mutations associated with polycystic kidney disease have played an important role in the advancement of ADPKD research. The Han:SPRD rat model, carrying an R823W mutation in the Anks6 gene is a well-documented animal model of inherited PKD with an autosomal dominant pattern of inheritance, closely mirroring several features of human ADPKD, including renal hyperplasia, azotemia, and extrarenal manifestations. The mutated protein, named Samcystin, is localized in cilia of tubular epithelial cells and seems to be involved in cystogenesis. The homozygous Anks6 mutation leads to end-stage renal disease and death, making it a critical factor in kidney development and function.
1. Introduction
2. The Han:SPRD Rat Model
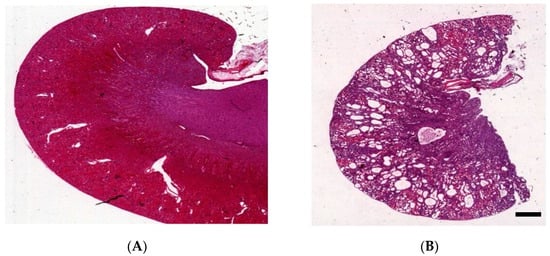
3. Kidney Histology of Han:SPRD Rats
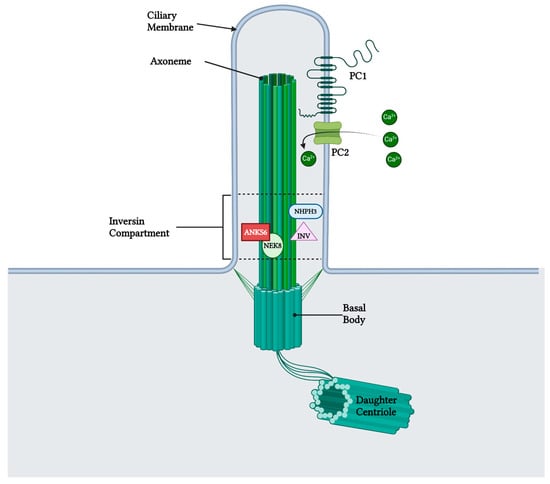
4. Genetics
5. Localization and Function of Samcystin in Kidney
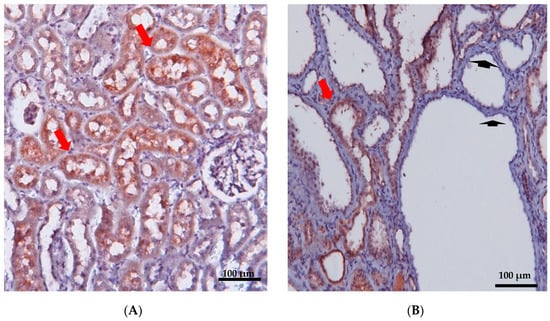
6. Role of Samcystin in Cystogenesis
7. Extrarenal Manifestations of ADPKD
8. Preclinical Trials
| Mechanism of Action |
Intervention | Han:SPRD | Human | ||
|---|---|---|---|---|---|
| Cyst Growth | Other Effect | Cyst Growth | Other Effect | ||
| mTOR inhibitor | Sirolimus | Reduction [47][48] | Reduction of CKD progression [47][48] | Reduction [49] | No effect in CKD progression [49] |
| COX-2 inhibitor | NS-398 | Reduction [50] | N/A | N/A | N/A |
| SGLT1,2i | Phlorizin | Reduction [51] | Reduction of CKD progression [51] | N/A | N/A |
| SGLT2i | Dapagliflozin | No effect [52][53][54] | Reduction of CKD progression [52][53][54] | Increase [55] | Increase of CKD progression [55] |
| Calcium channel inhibitors | Verapamil | Increase [56] | Increase of CKD progression [56] | N/A [57] | Increase of CKD progression [57] |
| Natural vitamin | Fish oil | None [58] | Reduction of diastolic dysfunction [58] | N/A | N/A |
9. ANKS6 in Humans
References
- Kurschat, C.E.; Müller, R.U.; Franke, M.; Maintz, D.; Schermer, B.; Benzing, T. An approach to cystic kidney diseases: The clinician’s view. Nat. Rev. Nephrol. 2014, 10, 687–699.
- Cramer, M.T.; Guay-Woodford, L.M. Cystic Kidney Disease: A Primer. Adv. Chronic Kidney Dis. 2015, 22, 297–305.
- Rohatgi, R. Extra-Renal Manifestations 5. Nephronophthisis (NPHP)-Medullary Cystic Kidney Disease (MCKD). 2008. Available online: http://pkdb.mayo.edu, (accessed on 31 January 2023).
- Iglesias, C.G.; Torres, V.E.; Offord, K.P.; Holley, K.E.; Beard, C.M.; Kurland, L.T. Epidemiology of Adult Polycystic Kidney Disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney Dis. 1983, 2, 630–639.
- Solazzo, A.; Testa, F.; Giovanella, S.; Busutti, M.; Furci, L.; Carrera, P.; Ferrari, M.; Ligabue, G.; Mori, G.; Leonelli, M.; et al. The prevalence of autosomal dominant polycystic kidney disease (ADPKD): A meta-analysis of European literature and prevalence evaluation in the Italian province of Modena suggest that ADPKD is a rare and underdiagnosed condition. PloS ONE 2018, 13, e0190430.
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Orskov, B.; Wanner, C.; Abad, J.M.; Aresté, N.; De La Torre, R.A.; Caskey, F.; Couchoud, C.; et al. Schaefer, Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: Prevalence and survival—An analysis of data from the ERA-EDTA Registry. Nephrol. Dial. Transplant. 2014, 29, iv15–iv25.
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal Dominant Polycystic Kidney Disease. 2007. Available online: http://www.thelancet.com (accessed on 15 February 2023).
- Guay-Woodford, L.M.; Henske, E.; Igarashi, P.; Perrone, R.D.; Reed-Gitomer, B.; Somlo, S.; Torres, V.E.; Ketchum, C.J.; Star, R.A.; Flessner, M.F.; et al. Filling the holes in cystic kidney disease research. Clin. J. Am. Soc. Nephrol. 2014, 9, 1799–1801.
- Torres, V.E.; Harris, P.C. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 18–32.
- Igarashi, P.; Somlo, S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398.
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710.
- Hanaoka, K.; Guggino, W.B. cAMP Regulates Cell Proliferation and Cyst Formation in Autosomal Polycystic Kidney Disease Cells. J. Am. Soc. Nephrol. 2000, 11, 1179–1187.
- Paul, B.M.; Heuvel, G.B.V. Kidney: Polycystic kidney disease. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 465–487.
- Halvorson, C.R.; Bremmer, M.S.; Jacobs, S.C. IJNRD-6939-Polycystic-Kidney-Disease—Inheritance—Pathology—Prognosis. 2010. Available online: https://www.dovepress.com/ (accessed on 25 February 2023).
- Hildebrandt, F.; Waldherr, R.; Kutt, R.; Brandis, M. Clinical Investigator The nephronophthisis complex: Clinical and genetic aspects. Clin. Investig. 1992, 70, 802–808.
- Omran, H.; Fernandez, C.; Jung, M.; Häffner, K.; Fargier, B.; Villaquiran, A.; Waldherr, R.; Gretz, N.; Brandis, M.; Rüschendorf, F.; et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am. J. Hum. Genet. 2000, 66, 118–127.
- Gupta, S.; Ozimek-Kulik, J.E.; Phillips, J.K. Nephronophthisis-pathobiology and molecular pathogenesis of a rare kidney genetic disease. Genes 2021, 12, 1762.
- Hildebrandt, F.; Zhou, W. Nephronophthisis-associated ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871.
- Ala-Mellol, S.; Kivivuorp, S.M.; Rrnnholm, K.A.R.; Koskimies, O.; Siimes, M.A. Mechanism underlying early anaemia in children with familial juvenile nephronophthisis. Pediatr. Nephrol. 1996, 10, 578–581.
- Waldherr, R.; Lennert, T.; Weber, H.; Schfirer, K.; Derks, H.; Rieger, P. rhiv A The Nephronophthisis Complex A Clinicopathologic Study in Children. Virchows Arch. A 1982, 394, 235–254.
- Parisi, M.A. Clinical and molecular features of Joubert syndrome and related disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 326–340.
- Baris, H.; Bejjani, B.A.; Tan, W.H.; Coulter, D.L.; Martin, J.A.; Storm, A.L.; Burton, B.K.; Saitta, S.C.; Gajecka, M.; Ballif, B.C.; et al. Identification of a novel polymorphism—The duplication of the NPHP1 (nephronophthisis 1) gene . Am. J. Med. Genet. Part A 2006, 140, 1876–1879.
- Guay-Woodford, L.M. Murine models of polycystic kidney disease: Molecular and therapeutic insights. Am. J. Physiol. Ren. Physiol. 2003, 285, F1034–F1049.
- Nagao, S.; Morita, M.; Kugita, M.; Yoshihara, D.; Yamaguchi, T.; Kurahashi, H.; Calvet, J.P.; Wallace, D.P. Polycystic kidney disease in Han:SPRD Cy rats is associated with elevated expression and mislocalization of SamCystin. Am. J. Physiol. Ren. Physiol. 2010, 299, 1078–1086.
- Kaspareit-Rittinghausen, J.; Rapp, K.; Deerberg, F.; Wcislo, A.; Messow, C. Hereditary Polycystic Kidney Disease Associated with Osteorenal Syndrome in Rats. Vet. Pathol. 1989, 26, 195–201.
- Bihoreau, M.-T.; Ceccherini, I.; Browne, J.; Kränzlin, B.; Romeo, G.; Lathrop, G.M.; James, M.R.; Gretz, N. Location of the First Genetic Locus, PKDr1, Controlling Autosomal Dominant Polycystic Kidney Disease in Han:SPRD cy/+ Rat; Oxford University Press: Oxford, UK, 1997.
- Ramasubbu, K.; Gretz, N.; Bachmannn, S. Increased Epithelial Cell Proliferation and Abnormal Extracellular Matrix in Rat Polycystic Kidney Disease. J. Am. Soc. Nephrol. 1998, 9, 937–945.
- Kränzlin, B.; Schieren, G.; Gretz, N. Azotemia and extrarenal manifestations in old female Han:SPRD (cy/+) rats. Kidney Int. 1997, 51, 1160–1169.
- Schafer, K.; Gretz, N.; Bader, M.; Oberbaumer, I.; Eckardt, K.-U.; Kriz, W.; Bachmann, S. Characterization of the Han:SPRD rat model for hereditary polycystic kidney disease. Kidney Int. 1994, 46, 134–152.
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal Models for Human Polycystic Kidney Disease. Exp. Anim. 2012, 61, 477–488.
- Kaspareit-Rittinghausen, J.; Deerberg, F.; Wcislo, A. Animal Model of Human Disease Hereditary Polycystic Kidney Disease Adult Polycystic Kidney Disease Associated with Renal Hypertension, Renal Osteodystrophy, and Uremic Enteritis in SPRD Rats. Am. J. Pathol. 1991, 139, 693.
- Nagao, S.; Yamaguchi, T.; Kusaka, M.; Maser, R.L.; Takahashi, H.; Cowley, B.D.; Grantham, J.J. Nagao, Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int. 2003, 63, 427–437.
- Cowley, B.D.; Gudapaty, S.; Kraybill, A.L.; Barash, B.D.; Harding, M.A.; Calvet, J.P.; Gattone, V.H. Autosomal-dominant polycystic kidney disease in the rat. Kidney Int. 1993, 43, 522–534.
- Kaisaki, P.J.; Bergmann, C.; Brown, J.H.; Outeda, P.; Lens, X.M.; Peters, D.J.M.; Gretz, N.; Gauguier, D.; Bihoreau, M.T. Genomic organization and mutation screening of the human ortholog of Pkdr1 associated with polycystic kidney disease in the rat. Eur. J. Med. Genet. 2008, 51, 325–331.
- Stagner, E.E.; Bouvrette, D.J.; Cheng, J.; Bryda, E.C. The polycystic kidney disease-related proteins Bicc1 and SamCystin interact. Biochem. Biophys. Res. Commun. 2009, 383, 16–21.
- Taskiran, E.Z.; Korkmaz, E.; Gucer, S.; Kosukcu, C.; Kaymaz, F.; Koyunlar, C.; Bryda, E.C.; Chaki, M.; Lu, D.; Vadnagara, K.; et al. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J. Am. Soc. Nephrol. 2014, 25, 1653–1661.
- Hoff, S.; Halbritter, J.; Epting, D.; Frank, V.; Nguyen, T.M.T.; Van Reeuwijk, J.; Boehlke, C.; Schell, C.; Yasunaga, T.; Helmstädter, M.; et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013, 45, 951–956.
- Bakey, Z.; Bihoreau, M.T.; Piedagnel, R.; Delestré, L.; Arnould, C.; De Villiers, A.D.; Devuyst, O.; Hoffmann, S.; Ronco, P.; Gauguier, D.; et al. The SAM domain of ANKS6 has different interacting partners and mutations can induce different cystic phenotypes. Kidney Int. 2015, 88, 299–310.
- Neudecker, S.; Walz, R.; Menon, K.; Maier, E.; Bihoreau, M.T.; Obermüller, N.; Kränzlin, B.; Gretz, N.; Hoffmann, S.C. Transgenic overexpression of Anks6(p.R823W) causes polycystic kidney disease in rats. Am. J. Pathol. 2010, 177, 3000–3009.
- Czarnecki, P.G.; Gabriel, G.C.; Manning, D.K.; Sergeev, M.; Lemke, K.; Klena, N.T.; Liu, X.; Chen, Y.; Li, Y.; Agustin, J.T.S.; et al. ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat. Commun. 2015, 6, 6023.
- Natoli, T.A.; Gareski, T.C.; Dackowski, W.R.; Smith, L.; Bukanov, N.O.; Russo, R.J.; Husson, H.; Matthews, D.; Piepenhagen, P.; Ibraghimov-Beskrovnaya, O. Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am. J. Physiol. Ren. Physiol. 2008, 294, F73–F83.
- Claus, L.R.; Chen, C.; Stallworth, J.; Turner, J.L.; Slaats, G.G.; Hawks, A.L.; Mabillard, H.; Senum, S.R.; Srikanth, S.; Flanagan-Steet, H.; et al. Certain heterozygous variants in the kinase domain of the serine/threonine kinase NEK8 can cause an autosomal dominant form of polycystic kidney disease. Kidney Int. 2023, 104, 995–1007.
- Wolf, M.T.F.; Hildebrandt, F. Nephronophthisis. Pediatr. Nephrol. 2011, 26, 181–194.
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41.
- Reiss, A.B.; Miyawaki, N.; Moon, J.; Kasselman, L.J.; Voloshyna, I.; D’Avino, R.; De Leon, J. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis 2018, 278, 49–59.
- Elias, R.M.; Dalboni, M.A.; Coelho, A.C.E.; Moysés, R.M.A. CKD-MBD: From the Pathogenesis to the Identification and Development of Potential Novel Therapeutic Targets. Curr. Osteoporos. Rep. 2018, 16, 693–702.
- Zafar, I.; Belibi, F.A.; He, Z.; Edelstein, C.L. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol. Dial. Transplant. 2009, 24, 2349–2353.
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wüthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2006, 21, 598–604.
- Liu, Y.M.; Shao, Y.Q.; He, Q. Sirolimus for treatment of autosomal-dominant polycystic kidney disease: A meta-analysis of randomized controlled trials. Transplant. Proc. 2014, 46, 66–74.
- Sankaran, D.; Bankovic-Calic, N.; Ogborn, M.R.; Crow, G.; Aukema, H.M. Selective COX-2 inhibition markedly slows disease progression and attenuates altered prostanoid production in Han:SPRD-cy rats with inherited kidney disease. Am. J. Physiol. Ren. Physiol. 2007, 293, F821–F830.
- Wang, X.; Zhang, S.; Liu, Y.; Spichtig, D.; Kapoor, S.; Koepsell, H.; Mohebbi, N.; Segerer, S.; Serra, A.L.; Rodriguez, D.; et al. Targeting of sodium-glucose cotransporters with phlorizin inhibits polycystic kidney disease progression in Han:SPRD rats. Kidney Int. 2013, 84, 962–968.
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE 2016, 11, e0146654.
- Rodriguez, D.; Kapoor, S.; Edenhofer, I.; Segerer, S.; Riwanto, M.; Kipar, A.; Yang, M.; Mei, C.; Wüthrich, R.P. Inhibition of Sodium-GlucoseCotransporter 2 with Dapagliflozin in Han: SPRD Rats with Polycystic Kidney Disease. Kidney Blood Press. Res. 2015, 40, 638–647.
- Afsar, B.; Afsar, R.E.; Demiray, A.; Altay, S.; Korkmaz, H.; Yildiz, A.; Covic, A.; Ortiz, A.; Kanbay, M. Sodium-glucose cotransporter inhibition in polycystic kidney disease: Fact or fiction. Clin. Kidney J. 2022, 15, 1275–1283.
- Morioka, F.; Nakatani, S.; Uedono, H.; Tsuda, A.; Mori, K.; Emoto, M. Short-Term Dapagliflozin Administration in Autosomal Dominant Polycystic Kidney Disease—A Retrospective Single-Arm Case Series Study. J. Clin. Med. 2023, 12, 6341.
- Nagao, S.; Nishii, K.; Yoshihara, D.; Kurahashi, H.; Nagaoka, K.; Yamashita, T.; Takahashi, H.; Yamaguchi, T.; Calvet, J.P.; Wallace, D.P. Calcium channel inhibition accelerates polycystic kidney disease progression in the Cy/+ rat. Kidney Int. 2008, 73, 269–277.
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shiohira, S.; Shimada, K.; Nitta, K.; Tsuchiya, K. Clinical effects of calcium channel blockers and renin-angiotensin- aldosterone system inhibitors on changes in the estimated glomerular filtration rate in patients with polycystic kidney disease. Clin. Exp. Nephrol. 2010, 14, 573–577.
- Ibrahim, N.H.M.; Thandapilly, S.J.; Jia, Y.; Netticadan, T.; Aukema, H. Soy Protein Alleviates Hypertension and Fish Oil Improves Diastolic Heart Function in the Han:SPRD-Cy Rat Model of Cystic Kidney Disease. Lipids 2016, 51, 635–642.
- Leettola, C.N.; Knight, M.J.; Cascio, D.; Hoffman, S.; Bowie, J.U. Characterization of the SAM domain of the PKD-related protein ANKS6 and its interaction with ANKS3. BMC Struct. Biol. 2014, 14, 17.
- Fang, B.; Guo, J.; Hao, C.; Guo, R.; Qian, S.; Li, W.; Jia, X. Whole-exome sequencing identifies a novel compound heterozygous mutation of ANKS6 gene in a Chinese nephronophthisis patient. Clin. Chim. Acta 2020, 501, 131–135.

