Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Brandon Lucke-Wold | -- | 4319 | 2024-02-28 17:50:47 | | | |
| 2 | Peter Tang | + 3 word(s) | 4322 | 2024-02-29 04:30:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Turner, R.C.; Lucke-Wold, B.; Lucke-Wold, N.; Elliott, A.S.; Logsdon, A.F.; Rosen, C.L.; Huber, J.D. Neuroprotection for Ischemic Stroke. Encyclopedia. Available online: https://encyclopedia.pub/entry/55675 (accessed on 13 June 2026).
Turner RC, Lucke-Wold B, Lucke-Wold N, Elliott AS, Logsdon AF, Rosen CL, et al. Neuroprotection for Ischemic Stroke. Encyclopedia. Available at: https://encyclopedia.pub/entry/55675. Accessed June 13, 2026.
Turner, Ryan C., Brandon Lucke-Wold, Noelle Lucke-Wold, Alisa S. Elliott, Aric F. Logsdon, Charles L. Rosen, Jason D. Huber. "Neuroprotection for Ischemic Stroke" Encyclopedia, https://encyclopedia.pub/entry/55675 (accessed June 13, 2026).
Turner, R.C., Lucke-Wold, B., Lucke-Wold, N., Elliott, A.S., Logsdon, A.F., Rosen, C.L., & Huber, J.D. (2024, February 28). Neuroprotection for Ischemic Stroke. In Encyclopedia. https://encyclopedia.pub/entry/55675
Turner, Ryan C., et al. "Neuroprotection for Ischemic Stroke." Encyclopedia. Web. 28 February, 2024.
Copy Citation
The translation of neuroprotective agents for ischemic stroke from bench-to-bedside has largely failed to produce improved treatments since the development of tissue plasminogen activator (tPA). One possible reason for lack of translation is the failure to acknowledge the greatest risk factor for stroke, age, and other common comorbidities such as hypertension, obesity, and diabetes that are associated with stroke.
ischemic stroke
comorbidity
aging
macrophage polarization
1. Introduction
1.1. Concept of Neuroprotection in Ischemic Stroke
Following the onset of ischemic stroke, cerebral blood flow is disrupted throughout the affected region of the brain. Blood flow disruption to the tissue is not uniform due to the presence of collateral circulation resulting in a flow gradient. Consequently, a “core” region of infarct develops in which flow is severely reduced, often approximating upwards of a 90% decrement, and tissue undergoes necrosis within minutes as insufficient adenosine triphosphate (ATP) is present to maintain homeostatic ionic gradients and metabolic functions [1]. Surrounding this core region in which blood flow is severely reduced is the “penumbra”, a tissue distribution in which flow is less severely reduced and remains typically around 35% of baseline flow [1]. Characterized by normal cellular membrane potential but a disruption in the ability for normal action potential firing, tissue initially residing within the penumbral region progresses to cellular death, ultimately resulting in an expanded core lesion without restoration of cerebral blood flow [2]. As such, therapeutic development has focused on achieving reperfusion via tissue plasminogen activator (tPA) and/or restoring homeostasis via administration of ‘neuroprotective’ pharmacologic agents intended to disrupt the ischemic injury cascade.
1.2. Current Status of Neuroprotectant Development: Drug Development Shortcomings
Investigation of neuroprotection and the associated concepts have gone through various periods of apparent focus as described by Lo previously [2]. Early studies, initiated by Astrup and colleagues, focused on understanding the effect of ischemia produced across a range of cerebral blood flow measurements on neurophysiology and electrical conduction [1]. The second phase of investigation sought to identify molecular mechanisms of injury with an emphasis on the acute period of injury. Following molecular biology studies was the discovery and implementation of numerous imaging modalities in both preclinical and clinical studies for assessing both changes in blood flow and the evolution of both the penumbra and infarct core [2]. While these various developments have provided much insight and utility in both preclinical and clinical studies, successful translation of a neuroprotectant from bench-to-bedside has yet to occur despite identification of over 1000 “successful” neuroprotectants in preclinical studies and initiation of over 100 clinical trials [3].
The reasons for lack of translation and failure are presently unknown but clear discrepancies exist between preclinical models and the clinical population most commonly afflicted by ischemic stroke. These include the age and general health of the population being studied as well as the time point of treatment initiation. Similarly, the endpoints frequently used in preclinical and clinical studies are often different. Furthermore, the vast majority of preclinical work has been conducted in rodents, species that are more distant phylogenetically from the human population in comparison to nonhuman primates [4]. Nonhuman primates more closely resemble humans in vascular anatomy, experience striatal damage following ischemic stroke more comparably to the human, and are gyrencephalic in nature like humans [4]. Consequently, nonhuman primates represent perhaps the most ideal preclinical model but are frequently not utilized due to cost, lack of availability, and expertise required for care and use. For these reasons, nonhuman primate models are not discussed further.
2. Improving Animal Models: Role of Comorbidities & Lifestyle
2.1. Accounting for the Aging Process: Effects on Inflammation
Age is the greatest risk factor for ischemic stroke yet has frequently not been emphasized in preclinical studies. Aging shifts the body to a pro-inflammatory state [5][6] leading to greater susceptibility to infection and injury as well as eventual immune exhaustion [7]. Aging also increases oxidative stress and immunosenescence [8]. Following stroke, an aged brain suffers greater blood brain barrier (BBB) disruption than a young brain [9]. The effects of aging are mediated through changes in astrocytes, macrophages, and microglia.
2.2. Effect of Comorbidities on Stroke Risk & Outcome
Comorbidities have long been known to increase risk for myocardial infarction and stroke. Until recently, individual diseases such as diabetes and hypertension were studied in isolation. Clinicians and researchers alike are now focusing on how multiple variables interact, and subsequently how they increase the threat for vascular disorder [10]. The metabolic syndrome is strongly associated with increased hazard risk for ischemic stroke [11]. It consists of hypertension, insulin resistance, obesity, and hypertriglyceridemia [12]. Although the causes of this syndrome are still under investigation, it is known that sedentary lifestyle, poor diet, and genetics play a role [13].
2.3. Accounting for Lifestyle Influences: Effects of Altered Sleep-Wake Patterns
In addition to the incorporation of comorbidities in ischemic stroke models, preclinical studies need to address the effect of variations in lifestyle that may lead to altered risk or outcome. An example of a lifestyle alteration that needs to be addressed is sleep deprivation and/or fragmentation. Sleep plays an integral role in everyday life, consuming approximately one third of a person’s day. Epidemiological studies have shown sleep to be important to a person’s health, with a positive association seen between sleep disruption and morbidity and mortality [14][15][16][17]. People who report having short sleep durations, durations of ≤6 h a night, have an increased prevalence of Type II diabetes, hypertension, obesity, cardiovascular disease, and stroke [18][19][20][21][22][23][24][25]. A study in Finland found that people engaged in shift work, defined as work outside regular daytime hours, had a higher incidence of stroke [26]. Sleep disruption is a characteristic of shift work, and people who are engaged in shift work are more likely to have shorter sleep durations than people who work regular daytime hours [27]. According to the 2010 National Health Interview Survey (NHIS), over 40 million employed U.S. adults report having short sleep durations. Therefore, the specific functions and molecular mechanisms of sleep are an active area of research.
In an effort to understand sleep, studies evaluate the consequences when sleep is taken away. In animal models, the function of sleep is analyzed predominantly by preventing the animal from sleeping, termed sleep deprivation (SD). Sleep fragmentation disrupts the quality of sleep without decreasing total sleep time and is less studied. Unfortunately, the method of sleep deprivation is highly variable within the literature and makes results problematic to interpret. Some SD methods place the rodent in a constantly rotating drum or on a platform (“disk-over-water” or DOW) where the animal must continuously move and cannot fall asleep [28][29]. Other methods place the rodent inside an automated running wheel that is activated when the animal falls asleep via electromyogram (EMG) monitoring [30]. The gentle handling method, where the animal is lightly poked or brushed when sleep is observed, is considered to be one of the least stressful methods for the animal [28]. However, this method is not automated and can be taxing on the researcher. Consequently, it is usually only used for acute SD studies of 6 hours or less. Duration of SD is also variable with acute durations of 4 or 6 h, chronic SD of 24–72 h, and repeated bouts of acute SD for days or weeks. This degree of variation makes the ability to interpret the results and generalize between studies problematic.
Within the stroke literature, acute SD prior to cerebral ischemia or injury has been shown to attenuate the severity of the injury, and could possibly be neuroprotective [31][32]. These findings are contradictory to epidemiological studies [26][25]. However, the sleep patterns in the animal studies have normal sleep and then experience shortened sleep duration prior to cerebral insult where the general population experience short sleep durations habitually. Consequently, studies evaluating the effects of sleep disruption on stroke risk and stroke severity need to more closely replicate the sleep patterns seen within the general population. Furthermore, the importance of sleep quality and continuity needs to be further evaluated since fragmented sleep is another characteristic of sleep disruption shown to have negative consequences [33]. Replicating the sleep patterns of the general public in animal studies will allow us to identify the mechanisms and signaling pathways taking place in the human population and discover where intervention is possible to reduce the poor health outcomes associated with sleep disruption.
2.4. Identification of Clinically-Relevant Endpoints for Preclinical Studies
In addition to the incorporation of animal models more consistent with the clinical population afflicted by stroke, whether that includes the various comorbidities or other lifestyle influences, preclinical models also likely need to emphasize functional or behavioral measures rather than strict histological analysis. Preclinical studies have long emphasized measurements of infarct volume in assessing therapeutic efficacy despite the fact that the emphasis clinically is on restoration of function and assessment of functional ability. In fact, a clinical study conducted by Saver and colleagues demonstrated that infarct volume serves as a relatively poor surrogate measure of functional outcome with subacute infarct volume displaying only a moderate correlation with 3-month clinical outcome [34]. Consequently, it is clear that preclinical studies may be improved by increasing the emphasis of functional outcome rather than infarct volume, a fact that has been recognized by leading advisory groups in the stroke community such as Stroke Therapy Academic and Industry Roundtable (STAIR) [35]. While multiple groups have performed infarct volume-function correlation studies in preclinical models, these models have employed young-adult animals rather than the potentially more clinically-relevant aged animals or those with comorbidities, limiting the ability to conclude whether volumetric measurements correlate and to what extent with functional capability. Other potentially relevant endpoints of use include the use of diagnostic imaging, such as magnetic resonance imaging (MRI), and cerebral blood flow measurements that are commonly used in the clinic. These may be particularly useful when performing longer duration studies to track infarct evolution over time and also to assess the therapeutic window of opportunity.
3. Promising Directions and Potential Targets in Neuroprotectant Development
Despite past shortcomings in neuroprotectant development, multiple pharmacologic agents are currently under investigation in advanced phases of clinical trial development and appear promising based on early phase results. This includes Arundic Acid (ONO-2506), an astrocyte modulator, and minocycline, an inhibitor of microglia.
3.1. Modulating Astrocyte Activity
While neuroprotection research has been largely neurocentric in nature and failed to emphasize the prominent role of glia in both homeostasis and injury response, the suggestion has been made that preservation of neuronal survival and function may not be possible without significant regulation of the microenvironment by astrocytes [36]. Astrocytes, frequently described as “housekeeping cells of the nervous system”, are involved in regulating blood-brain barrier (BBB) integrity, removal of metabolic waste products, release of neural growth factors, and uptake of excess neurotransmitter release during synaptic activity [36]. As such, astrocyte modulation represents a promising strategy for future therapeutic development for ischemic stroke [36]. The effect of ischemia on astrocytes is widespread in that signaling amongst astrocytes, as well as between astrocytes and neurons or microglial cells, is likely altered in addition to the potential for necrotic or apoptotic cellular death. This disrupted signaling likely has significant consequences on the control of basic physiologic functions including cerebral blood flow, glutamate uptake by astrocytes, and the ability of astrocytes to serve as an energy reserve during ischemia [37][38][39][40].
Perhaps the hallmark event involving astrocytes following neural injury, and specifically ischemia, is the development of reactive gliosis and associated morphologic changes and altered gene expression [41]. One of the challenges in modulating astrocyte activity, and associated gliosis, for therapeutic benefit is the emerging evidence that astrocytes have both protective and detrimental roles, potentially a result of multiple astrocyte subtypes [41]. Recent work by Zamanian, et al. have illustrated this concept in that ischemia appears to promote a protective astrocytic response whereas neuroinflammation induced by lipopolysaccharide (LPS) produces a potentially detrimental phenotype. Notably, these two different stimuli each induced significantly altered gene expression yet the expression shared between stimuli was less than 50%, furthering the notion of a stimulus-specific astrocyte response or multiple astrocyte subtypes [41]. The influence of astrocytes on outcome following neurologic injury, and the potential for conflicting roles (beneficial or detrimental), has been seen clearly in a variety of preclinical models including ischemia, experimental autoimmune encephalitis (EAE), and spinal cord injury to name a few. In a preclinical model of ischemia, Li and colleagues demonstrated a protective role of astrocytes by knocking out glial fibrillary acidic protein (GFAP) and vimentin [42]. Deletion of this combination of intermediate filaments, characteristic of astrocytes, resulted in infarct volumes two to three times larger than in wild-type animals [42]. In spinal cord injury, a dual role of astrocyte reactivity has been observed in which initially, astrocytes appear to modulate inflammation and contain inflammatory cells to the damaged region. On the other hand, astrocytes are the primary component of the glial scar that prevents axonal re-growth in the chronic period post-injury [43]. In a model of demyelinating disease, experimental autoimmune encephalitis (EAE), targeted genetic removal of reactive astrocytes resulted in diminished perivascular scar formation and increased leukocyte infiltration into brain parenchyma [44]. The exacerbated inflammatory response was associated with a detrimental outcome [44].
Despite the expanded understanding of astrocyte roles in homeostasis and disease, clear knowledge gaps remain. This includes more clear elucidation of the timing of beneficial versus detrimental responses to injury, how these responses can be pharmacologically manipulated, and how the astrocyte response is altered in association with disease comorbidities and the aging process. The researchers' laboratory and others have begun to explore these factors with the hope of improving the understanding of neural injury in order to develop future therapeutics [45][46].
One potentially promising therapeutic that alters astrocyte activation, and synthesis of S-100B, that has advanced to clinical trials is (R)-(–)-2-propyloctanoic acid (ONO-2506). S-100B is expressed by reactive astrocytes throughout the penumbral region following ischemia and leads to nitric oxide release from the astrocyte, potentiating neuronal cell death [47][48]. In an early phase clinical trial, ONO-2506, also known as arundic acid, proved safe and produced a trend towards functional improvement based on the National Institutes of Health Stroke Scale (NIHSS) [47].
3.2. Inhibiting Effects of Microglia
Much like astrocytes, microglial modulation may represent a promising therapeutic target for ischemic stroke based on the progression of compounds such as minocycline in clinical trials. Microglial activation, a hallmark of the inflammatory response within the central nervous system post-stroke, has been described as both beneficial and detrimental in nature [49]. Ablation of Mac-2 positive microglia, a marker of activated microglia, resulted in enhanced infarct volume and altered temporality of pro-inflammatory cytokine release. These changes were associated with a nearly three-fold increase in apoptotic cells and a nearly two-fold decrease in IGF-1, a neurotrophic factor, levels [50]. In contrast, numerous other studies have shown an improvement in outcome following ischemia by reducing microglial activation [51][52]. This is achieved by administration of minocycline, traditionally used as an antibiotic, which has been associated with diminished Iba-1 expression, an activation marker, by microglia [51]. Importantly, the microglial response post-stroke begins rapidly but takes hours to days to develop fully, potentially resulting in an extended window of therapeutic opportunity [49]. The idea of an extended window, at least in comparison to that used for thrombolytic therapy, is consistent with preclinical findings in which a clinically relevant dosage of minocycline decreased infarct volume at both 4 and 5 h post-stroke while reducing functional deficits with 4 h administration [53]. In addition to the effects on microglia, minocycline has been shown to alter the function of other immune cells previously implicated in ischemic damage such as macrophages [54]. While not previously investigated specifically with regards to ischemia, the effect of minocycline on macrophages may be related to alterations in macrophage recruitment and/or macrophage polarization, a topic that is discussed at greater length in subsequent sections. The effect of minocycline on subsequent inflammatory processes is logical in that microgliosis following injury is associated with not only the acute and rapid activation of microglial cells but also the recruitment of marrow-derived inflammatory cells that migrate to the brain and penetrate the parenchyma [50]. The precise mechanism via which minocycline mediates protection through microglial-mediated effects remains unclear but has been shown to decrease expression of high-mobility group protein B1 (HMGB1) and in vitro, reduces oxidative stress via diminished superoxide release [51][55]. Minocycline may also prove beneficial in other reparative therapies post-stroke, such as stem-cell delivery. Sakata and colleagues demonstrated that minocycline pre-conditioning of neural stem cells prior to transplantation resulted in production of beneficial paracrine factors and diminished graft cell death via upregulation of antioxidant genes associated with Nrf2 [52].
In early phase clinical trials, minocycline has exhibited similar promise in that it has been proven safe when administered alone or in combination with thrombolytics and also exhibited potential efficacy in these small studies. Namely, NIHSS and modified Rankin Scale (mRS) scores were lower, and Barthel Index (BI) scores higher, in the group given minocycline [56]. This improvement was seen as soon as 7 days post-stroke and persisted at day 30 of follow-up [56]. While a large efficacy trial is needed to confirm preliminary findings, minocycline appears promising for the treatment of ischemic stroke [57]. Notably, clinical studies have also shown that minocycline administration results in diminished levels of matrix metalloproteinase-9 (MMP-9) [58]. Preclinical studies have shown a reduction in blood-brain barrier (BBB) permeability following minocycline administration and due to previous associations between BBB permeability and MMP-9 activity, it would appear that these clinical findings indicate a potential effect of minocycline on restoration or maintenance of BBB integrity [55][58].
3.3. Modulating the Blood-Brain Barrier (BBB)
As focus has gradually shifted from solely the neuron to include glial cells and the associated neurovascular unit, the role of the blood-brain barrier in ischemic injury has received renewed interest. Much of the work involving BBB permeability has focused on potential mechanisms of degradation, such as matrix metalloproteinases, creating a natural target for pharmacologic agents [59][60][61][62][63]. In work by Pfefferkorn and colleagues, the authors showed the ability to reduce BBB disruption and improve survival following delayed tPA administration by using BB-94, a matrix metalloproteinase inhibitor [64]. In more recent work using a model of traumatic brain injury, a similar phenomenon was shown using Kollidon VA64, a pharmacological agent described as resealing membranes [65]. Improving membrane integrity resulted in diminished neural injury and improved functional outcome [65]. It is clear that more investigation is required to understand not only the role of BBB permeability in disease but also how to target BBB permeability therapeutically, but studies such as those described herein provide a potential proof-of-concept and perhaps most importantly, further indicate the need for a comprehensive approach to ischemic stroke therapeutic development.
4. Targeting Inflammation
As demonstrated by the early phase success of agents currently under investigation, such as minocycline and arundic acid, targeting inflammation may offer promise in the quest for identification of neuroprotective compounds. Inflammation offers many opportunities and targets for modulation, many of which persist for hours to days, sometimes even longer, following injury. Consequently, these targeted therapeutics are more likely to be administered within the window of opportunity for a given pathologic process compared to some of the previously tested agents that target events occurring almost immediately post-stroke. Inflammation, while generally thought of in a negative connotation, is an essential component of recovery and repair-associated processes as well, further complicating experimental investigations.
4.1. Inflammation: A Deleterious Event or a Beneficial Response?
The inflammatory process is further complicated due to the fact that various elements can serve a deleterious or a beneficial role in the injury and recovery process, as highlighted earlier in this research with discussion of astrocytes and microglia. Another prime example of this potentially dual response to neural injury is that of macrophages. Macrophages and microglia may contribute to secondary injury processes via production of inflammatory cytokines and reactive oxygen species while also potentially reducing injury through maintenance of the cellular microenvironment by phagocytic clearance of cellular debris and apoptotic cells [66][67][68]. Emerging evidence indicates that many of these inflammatory cell types may never truly exist in a resting state and rather may almost constantly be undergoing changes in state through intermediary forms, evident by both morphologic and functional changes, in response to the cellular environment and signals from the surrounding regions [67]. This is perhaps most adequately described, and potentially targeted therapeutically, by macrophages. In the following section, the researchers attempt to highlight some of the recent developments in the macrophage literature, particularly those related to various macrophage subsets and neural injury.
4.2. Introduction to Macrophage Polarization
Macrophages have long been recognized as a key element in the inflammatory response, particularly in reference to invasion of the body by foreign material such as bacteria. Besides their role as immune effector cells, macrophages are perhaps most important for normal homeostatic processes such as cellular turnover and clearance of cellular debris, hence the description of macrophages as the “janitorial” cell of the body [69]. For example, macrophages are intimately involved in the clearance of over 200 billion erythrocytes each day as well as clearance of apoptotic cells [69]. Consequently, macrophages respond to both endogenous stimuli produced through injury as well as to foreign cell types [69]. As such, work in both the nervous system as well as other body systems indicates that macrophages can be shifted to either a pro-inflammatory or anti-inflammatory state depending on disease state and environmental cues [66]. Gaining a better understanding of the processes mediating this phenotypic shift may lead to therapeutic advances [66]. Perhaps the greatest challenge in this process is the complexity of elucidating these mechanisms in vivo. In vitro studies allow for rapid stimulation of inflammatory cells with antigens or cytokines but this response often does not translate to in vivo work, or is at the very least, often modified in a significant way [70].
4.3. M1 versus M2: Classical versus Alternative Activation
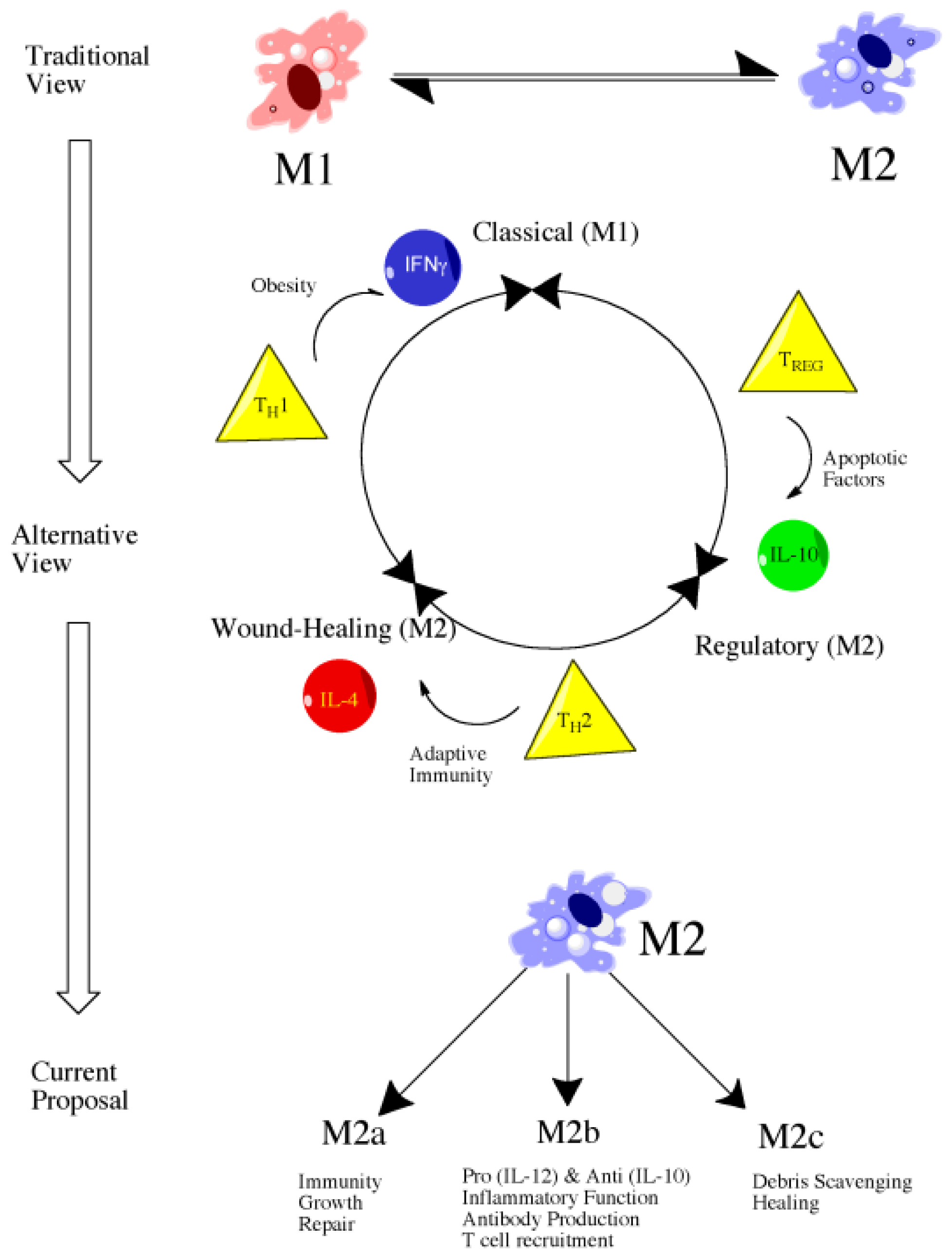
Literature focusing on macrophage polarization and subtypes has followed a similar path to that of T-cell literature and, consequently, macrophages have largely been classified as M1 or M2, a description that persists despite evidence showing greater complexity, particularly amongst the M2 population (Figure 1) [69]. In spite of the potential for additional subtypes, the researchers use the conventional M1 and M2 descriptions for organizational purpose but do explore the potential for various M2 subtypes within this research.
Figure 1. Evolution of views of macrophage polarization from the more simplistic M1 or M2 characterization without subtypes to the alternative or developing view with the M2 population divided into “wound-healing” and “regulatory” subtypes. Even more current proposals identify three populations of M2-related macrophages involved in a range of functions.
4.4. Classically Activated Macrophages (M1)
Classical macrophage activation refers to those designated as effector cells as part of cell-mediated immune responses. In the past, classical activation occurred in response to two primary signals—interferon gamma (IFNγ) and tumor necrosis factor (TNF) [69]. This results in increased secretion of pro-inflammatory cytokines and mediators such as IL-12, IL-23, IL-1β, TNF-α, reactive oxygen species (ROS), and nitrosylated species (NS), ultimately producing increased microbicidal or tumoricidal capability [66]. While emerging evidence indicates that other signals may also induce an M1 response, such as certain toll-like receptor (TLR) agonists, the idea remains the same in that M1 macrophages, while vital for host defense, must be carefully regulated due to the possibility of host-tissue damage with unchecked activation [69]. Persistent inflammation and the associated host-tissue damage is a hallmark of numerous autoimmune diseases such as rheumatoid arthritis and inflammatory bowel disease, demonstrating the need for regulation and strict control of activation processes [69].
4.5. Alternatively Activated Macrophages (M2)
Alternative macrophage activation, similar to classical macrophage activation, can occur in response to both innate and adaptive signals [69]. The primary signaling molecule responsible for activation is interleukin-4 (IL-4), a cytokine released upon initial tissue injury as well as upon initiation of the adaptive immune response. Alternatively activated macrophages are largely described as beneficial, particularly when it comes to wound healing. Specifically, alternative macrophage activation is responsible for production of extracellular matrix, a property essential in the reparative response. Alternatively activated macrophages are also associated with a reduced expression of pro-inflammatory cytokines, namely TNFα, IL-1β, IL-2, IL-8, IL-12, and CXCL10 [66]. The response of IL-4 stimulated alternatively activated macrophages is much more characteristic of a TH2-mediated response in comparison to classical macrophage activation which more closely parallels that of a TH1 response [66]. Despite the previous characterization of alternatively activated macrophages as a distinct cellular population, emerging evidence indicates the presence of further subtypes under the M2 umbrella and that they are involved in a variety of functions ranging from wound-healing to regulatory functions. In fact, some have described M2 macrophages as actually three distinct subtypes—M2a, M2b, and M2c, each of which is generated through a unique polarization process and performs distinct functions (Figure 1) [66]. M2a and M2c have been attributed clear anti-inflammatory and reparative roles whereas M2b are somewhat unique in that they produce high amounts of IL-10, an anti-inflammatory cytokine, but low-levels of IL-12, a pro-inflammatory cytokine [66]. Similarly, M2b macrophages may produce TNFα, IL-1β, and IL-6, indicating a more complex role in the inflammatory response than previously recognized [66].
4.6. Future Directions: Therapeutic Targeting of Macrophage Subsets
Therapeutically, it is clear that shifting from the pro-inflammatory macrophage phenotype, M1, to the anti-inflammatory phenotype, M2, may confer an advantage in both initial injury and long-term recovery. How precisely to alter the phenotype and at what time post-injury is not clear but the idea is promising, particularly considering that recent studies have demonstrated that macrophage subsets may not be truly distinct entities and rather may undergo switching. This idea has been demonstrated in vitro in which activating macrophages with LPS resulted in diminished reactivity to subsequent pro-inflammatory stimuli [71]. In contrast, these macrophages maintained the ability to express other genes associated with anti-inflammatory activities such as IL-10 [71]. The ability to diminish responsiveness to pro-inflammatory stimuli while maintaining the anti-inflammatory capabilities is referred to as endotoxic tolerance and is generally associated with a switch from the M1 to M2 phenotype [71].
While inflammation is clearly a promising therapeutic target, the understanding of inflammatory processes and when precisely each event occurs remains somewhat unclear and likely varies across individuals. As such, a need exists for not only improved measurement and insight into inflammatory processes but also a personalized approach, a common theme discussed throughout medical research in recent years. One method for potentially achieving both these goals is the use of blood-based or CSF-based biomarkers.
References
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 1977, 8, 51–57.
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med 2008, 14, 497–500.
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1026 experimental treatments in acute stroke. Ann. Neurol 2006, 59, 467–477.
- Fukuda, S.; del Zoppo, G.J. Models of focal cerebral ischemia in the nonhuman primate. ILAR J 2003, 44, 96–104.
- Bonafe, M.; Storci, G.; Franceschi, C. Inflamm-aging of the stem cell niche: Breast cancer as a paradigmatic example: Breakdown of the multi-shell cytokine network fuels cancer in aged people. Bioessays 2012, 34, 40–49.
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord. Drug Targets 2011, 10, 621–634.
- Simpson, R.J.; Lowder, T.W.; Spielmann, G.; Bigley, A.B.; LaVoy, E.C.; Kunz, H. Exercise and the aging immune system. Ageing Res. Rev 2012, 11, 404–420.
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflamm-aging and immunosenescence. J. Proteomics 2011, 74, 2313–2323.
- DiNapoli, V.A.; Huber, J.D.; Houser, K.; Li, X.; Rosen, C.L. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 2008, 29, 753–764.
- Goodman, E.; Li, C.; Tu, Y.K.; Ford, E.; Sun, S.S.; Huang, T.T. Stability of the factor structure of the metabolic syndrome across pubertal development: Confirmatory factor analyses of three alternative models. J. Pediatr 2009, 155, e1–e8.
- Chien, K.L.; Hsu, H.C.; Sung, F.C.; Su, T.C.; Chen, M.F.; Lee, Y.T. Metabolic syndrome as a risk factor for coronary heart disease and stroke: An 11-year prospective cohort in Taiwan community. Atherosclerosis 2007, 194, 214–221.
- Maruyama, K.; Uchiyama, S.; Iwata, M. Metabolic syndrome and its components as risk factors for first-ever acute ischemic noncardioembolic stroke. J. Stroke Cerebrovasc. Dis 2009, 18, 173–177.
- Lambert, G.W.; Straznicky, N.E.; Lambert, E.A.; Dixon, J.B.; Schlaich, M.P. Sympathetic nervous activation in obesity and the metabolic syndrome—Causes, consequences and therapeutic implications. Pharmacol. Ther 2010, 126, 159–172.
- Ayas, N.T.; White, D.P.; Manson, J.E.; Stampfer, M.J.; Speizer, F.E.; Malhotra, A.; Hu, F.B. A prospective study of sleep duration and coronary heart disease in women. Arch. Intern. Med 2003, 163, 205–209.
- Ferrie, J.E.; Shipley, M.J.; Cappuccio, F.P.; Brunner, E.; Miller, M.A.; Kumari, M.; Marmot, M.G. A prospective study of change in sleep duration: Associations with mortality in the Whitehall II cohort. Sleep 2007, 30, 1659–1666.
- Qureshi, A.I.; Giles, W.H.; Croft, J.B.; Bliwise, D.L. Habitual sleep patterns and risk for stroke and coronary heart disease: A 10-year follow-up from NHANES I. Neurology 1997, 48, 904–911.
- Ikehara, S.; Iso, H.; Date, C.; Kikuchi, S.; Watanabe, Y.; Wada, Y.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Association of sleep duration with mortality from cardiovascular disease and other causes for Japanese men and women: The JACC study. Sleep 2009, 32, 295–301.
- Sabanayagam, C.; Shankar, A. Sleep duration and cardiovascular disease: Results from the national health interview survey. Sleep 2010, 33, 1037–1042.
- Cappuccio, F.; Taggart, F.M.; Kandala, N.B.; Currie, A.; Peile, E.; Stranges, S.; Miller, M.A. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 2008, 31, 619–626.
- Chaput, J.P.; Despres, J.P.; Bouchard, C.; Tremblay, A. Association of sleep duration with type 2 diabetes and impaired glucose tolerance. Diabetologia 2007, 50, 2298–2304.
- Schultes, B.; Schmid, S.; Peters, A.; Born, J.; Fehm, H.L. Sleep loss and the development of diabetes: A review of current evidence. Exp. Clin. Endocrinol. Diabetes 2005, 113, 563–567.
- Spiegel, K.; Knutson, K.; Leproult, R.; Tasali, E.; van Cauter, E. Sleep loss: A novel risk factor for insulin resistance and Type 2 diabetes. J. Appl. Physiol 2005, 99, 2008–2019.
- Kim, J.; Jo, I. Age-dependent association between sleep duration and hypertension in the adult Korean population. Am. J. Hypertens 2010, 23, 1286–1291.
- Gottlieb, D.J.; Redline, S.; Nieto, F.J.; Baldwin, C.M.; Newman, A.B.; Resnick, H.E.; Punjabi, N.M. Association of usual sleep duration with hypertension: The sleep heart health study. Sleep 2006, 29, 1009–1014.
- Chen, J.C.; Brunner, R.L.; Ren, H.; Wassertheil-Smoller, S.; Larson, J.C.; Levine, D.W.; Allison, M.; Naughton, M.J.; Stefanick, M.L. Sleep duration and risk of ischemic stroke in postmenopausal women. Stroke 2008, 39, 3185–3192.
- Nurminen, M.; Karjalainen, A. Epidemiologic estimate of the proportion of fatalities related to occupational factors in Finland. Scand. J. Work Environ. Health 2001, 27, 161–213.
- Luckhaupt, S.E.; Tak, S.; Calvert, G.M. The prevalence of short sleep duration by industry and occupation in the national health interview survey. Sleep 2010, 33, 149–159.
- Fenzl, T.; Romanowski, C.P.; Flachskamm, C.; Honsberg, K.; Boll, E.; Hoehne, A.; Kimura, M. Fully automated sleep deprivation in mice as a tool in sleep research. J. Neurosci. Methods 2007, 166, 229–235.
- Rechtschaffen, A.; Bergmann, B.M.; Gilliland, M.A.; Bauer, K. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep 1999, 22, 11–31.
- Coenen, A.M.; van Luijtelaar, E.L. Stress induced by three procedures of deprivation of paradoxical sleep. Physiol. Behav 1985, 35, 501–504.
- Hsu, J.C.; Lee, Y.S.; Chang, C.N.; Ling, E.A.; Lan, C.T. Sleep deprivation prior to transient global cerebral ischemia attenuates glial reaction in the rat hippocampal formation. Brain Res 2003, 984, 170–181.
- Moldovan, M.; Constantinescu, A.O.; Balseanu, A.; Oprescu, N.; Zagrean, L.; Popa-Wagner, A. Sleep deprivation attenuates experimental stroke severity in rats. Exp. Neurol 2010, 222, 135–143.
- Bonnet, M.H.; Arand, D.L. Clinical effects of sleep fragmentation versus sleep deprivation. Sleep Med. Rev 2003, 7, 297–310.
- Saver, J.L.; Johnston, K.C.; Homer, D.; Wityk, R.; Koroshetz, W.; Truskowski, L.L.; Haley, E.C. Infarct volume as a surrogate or auxiliary outcome measure in ischemic stroke clinical trials. The RANTTAS Investigators. Stroke 1999, 30, 293–298.
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758.
- Takano, T.; Oberheim, N.; Cotrina, M.L.; Nedergaard, M. Astrocytes and ischemic injury. Stroke 2009, 40, S8–S12.
- Bambrick, L.; Kristian, T.; Fiskum, G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem. Res 2004, 29, 601–608.
- Carmignoto, G.; Gomez-Gonzalo, M. The contribution of astrocyte signalling to neurovascular coupling. Brain Res. Rev 2010, 63, 138–148.
- Rossi, D.J.; Brady, J.D.; Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci 2007, 10, 1377–1386.
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.A.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci 2003, 6, 43–50.
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci 2012, 32, 6391–6410.
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Stahlberg, A.; Aprico, K.; Larsson, K.; Yabe, T.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab 2008, 28, 468–481.
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med 2006, 12, 829–834.
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.; Tiwari-Woddruff, S.; Sofroniew, M.V. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci 2009, 29, 11511–11522.
- Cotrina, M.L.; Nedergaard, M. Astrocytes in the aging brain. J. Neurosci. Res 2002, 67, 1–10.
- Dinapoli, V.A.; Benkovic, S.A.; Li, X.; Kelly, K.A.; Miller, D.B.; Rosen, C.L.; Huber, J.D.; O’Callaghan, J.P. Age exaggerates proinflammatory cytokine signaling and truncates signal transducers and activators of transcription 3 signaling following ischemic stroke in the rat. Neuroscience 2010, 170, 633–644.
- Pettigrew, L.C.; Kasner, S.E.; Albers, G.W.; Gorman, M.; Grotta, J.C.; Sherman, D.G.; Funakoshi, Y.; Ishibashi, H. Arundic Acid (ONO-2506); Stroke Study Group. Safety and tolerability of arundic acid in acute ischemic stroke. J. Neurol. Sci. 2006, 251, 50–56.
- Tateishi, N.; Mori, T.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Matsui, T.; Asano, T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)-(–)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J. Cereb. Blood Flow Metab 2002, 22, 723–734.
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391.
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci 2007, 27, 2596–2605.
- Hayakawa, K.; Mishima, K.; Nozako, M.; Hazekawa, M.; Mishima, S.; Fujioka, M.; Orito, K.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke 2008, 39, 951–958.
- Sakata, H.; Niizuma, K.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Narasimhan, P.; Maier, C.M.; Nishiyama, Y.; Chan, P.H. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci 2012, 32, 3462–3473.
- Xu, L.; Fagan, S.C.; Waller, J.L.; Edwards, D.; Borlongan, C.V.; Zheng, J.; Hill, W.D.; Feuerstein, G.; Hess, D.C. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol 2004, 4, 7.
- Campbell, J.H.; Burdo, T.H.; Autissier, P.; Bombardier, J.P.; Westmoreland, S.V.; Soulas, C.; Gonzalez, R.G.; Ratai, E.-M.; Williams, K.C. Minocycline inhibition of monocyte activation correlates with neuronal protection in SIV neuroAIDS. PLoS One 2011, 6, e18688.
- Yenari, M.A.; Xu, L.; Tang, X.N.; Qiao, Y.; Giffard, R.G. Microglia potentiate damage to blood-brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke 2006, 37, 1087–1093.
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410.
- Fagan, S.C.; Cronic, L.E.; Hess, D.C. Minocycline development for acute ischemic stroke. Transl. Stroke Res 2011, 2, 202–208.
- Switzer, J.A.; Hess, D.C.; Ergul, A.; Waller, J.L.; Machado, L.S.; Dobos-Portik, V.; Pettigrew, L.C.; Clark, W.M.; Fagan, S.C. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke 2011, 42, 2633–2635.
- Lee, S.R.; Lo, E.H. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. J. Cereb. Blood Flow Metab 2004, 24, 720–727.
- Lee, S.R.; Tsuji, K.; Lo, E.H. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J. Neurosci 2004, 24, 671–678.
- Lo, E.H.; Wang, X.; Cuzner, M.L. Extracellular proteolysis in brain injury and inflammation: Role for plasminogen activators and matrix metalloproteinases. J. Neurosci. Res 2002, 69, 1–9.
- Rosell, A.; Lo, E.H. Multiphasic roles for matrix metalloproteinases after stroke. Curr. Opin. Pharmacol 2008, 8, 82–89.
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med 2006, 12, 441–445.
- Pfefferkorn, T.; Rosenberg, G.A. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke 2003, 34, 2025–2030.
- Mbye, L.H.; Keles, E.; Tao, L.; Zhang, J.; Chung, J.; Larvie, M.; Koppula, R.; Lo, E.H.; Whalen, M.J. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab 2012, 32, 515–524.
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci 2011, 12, 388–399.
- Polazzi, E.; Monti, B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol 2010, 92, 293–315.
- Popovich, P.G.; Longbrake, E.E. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci 2008, 9, 481–493.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 2008, 8, 958–969.
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol 2011, 11, 723–737.
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol 2011, 11, 750–761.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
507
Revisions:
2 times
(View History)
Update Date:
29 Feb 2024
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No