Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Michelerio | -- | 1775 | 2024-02-25 11:54:45 | | | |
| 2 | Peter Tang | Meta information modification | 1775 | 2024-02-26 03:11:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Michelerio, A.; Tomasini, C. Pathogenesis of Galli–Galli Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/55412 (accessed on 26 July 2026).
Michelerio A, Tomasini C. Pathogenesis of Galli–Galli Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/55412. Accessed July 26, 2026.
Michelerio, Andrea, Carlo Tomasini. "Pathogenesis of Galli–Galli Disease" Encyclopedia, https://encyclopedia.pub/entry/55412 (accessed July 26, 2026).
Michelerio, A., & Tomasini, C. (2024, February 25). Pathogenesis of Galli–Galli Disease. In Encyclopedia. https://encyclopedia.pub/entry/55412
Michelerio, Andrea and Carlo Tomasini. "Pathogenesis of Galli–Galli Disease." Encyclopedia. Web. 25 February, 2024.
Copy Citation
Galli–Galli disease (GGD) is a rare genodermatosis that exhibits autosomal dominant inheritance with variable penetrance. GGD typically manifests with erythematous macules, papules, and reticulate hyperpigmentation in flexural areas. A distinct atypical variant exists, which features brown macules predominantly on the trunk, lower limbs, and extremities, with a notable absence of the hallmark reticulated hyperpigmentation in flexural areas.
hyperpigmentation
acantholysis
keratin-5
skin diseases
genetic
papulosquamous
1. Introduction
Galli–Galli Disease (GGD) is a rare and poorly understood genodermatosis that exhibits an autosomal dominant inheritance pattern with variable penetrance [1][2], although sporadic cases lacking family history have been described [1]. First described by Bardach, Gebhart, and Luger in 1982 [3], GGD was identified in two brothers and has since been classified as part of the reticulate pigmented disorders of the skin (RPDS) group [4].
Clinically, GGD is characterized by erythematous macules and papules with reticulate hyperpigmentation affecting the flexural areas, including the axillary, inguinal, and neck regions. During the inflammatory phase of the disease, itchy, erythematous, vesicular, and/or scaly papules may manifest in the same distribution, as well as on the trunk and proximal extremities [5][6][7]. Comedo-like lesions or pitted acneiform scars on the face may occasionally occur [6][8][9]. An emerging consensus posits a distinct GGD subset characterized by an atypical phenotype. While histopathological features remain consistent with classical GGD, this variant diverges in its presentation of brown macules on the trunk, lower limbs, and extremities, almost absent the hallmark reticulated hyperpigmentation in flexural areas [5].
The clinical phenotype of classic GGD bears a striking resemblance to Dowling-Degos disease (DDD), another rare autosomal dominant genodermatosis characterized by reticulate pigmentary macules of the flexures, comedo-like lesions on the back, neck, and face, and perioral pitted acneiform scars [10][11]. In addition, there have been various reports in the literature that include atypical presentations of DDD or additional uncommon findings [10]. This significant overlap between GGD and DDD has led to ongoing confusion and misdiagnosis [12]. Traditionally, the differentiation has been primarily histopathologic, with suprabasal acantholysis serving as a distinctive feature that is historically attributable to GGD, thereby categorizing it as an acantholytic variant of DDD [5]. Finally, molecular genetic analyses have identified mutations in the KRT5 and POGLUT1 genes in some cases of both GGD and DDD; therefore, GGD is now referred to as a variant of DDD [7].
Despite the fact that GGD and DDD have very similar clinical aspects to each other, GGD has traditionally been considered to be a separate entity from DDD because suprabasal acantholysis is histopathologically observed only in GGD [12]. Advances in genetics have identified key mutations in genes such as KRT5 and POGLUT1 as pivotal in the pathogenesis of GGD. Intriguingly, these mutations have also been reported in cases of DDD [7][13].
2. KRT5
Keratin 5 (KRT5) protein coding gene, which was initially identified in DDD in 2006, is the main gene involved in the pathogenesis of GGD [8]. This gene is located on chromosome 12 (12q13.13), contains nine exons, and is predominantly expressed within the basal cells of the epidermis [14]. Its alteration, which is mainly caused by frameshift mutations, causes premature stop codons and results in haploinsufficiency (an insufficient production of the gene product) [5][14].
KRT5 is a protein that forms intermediate filaments, or heterodimers, with cytokeratin 14 (KRT14) [14][15]. Both proteins play a critical role in attaching keratinocytes together and anchoring the epidermis to the dermis (Figure 1) [14][15][16].
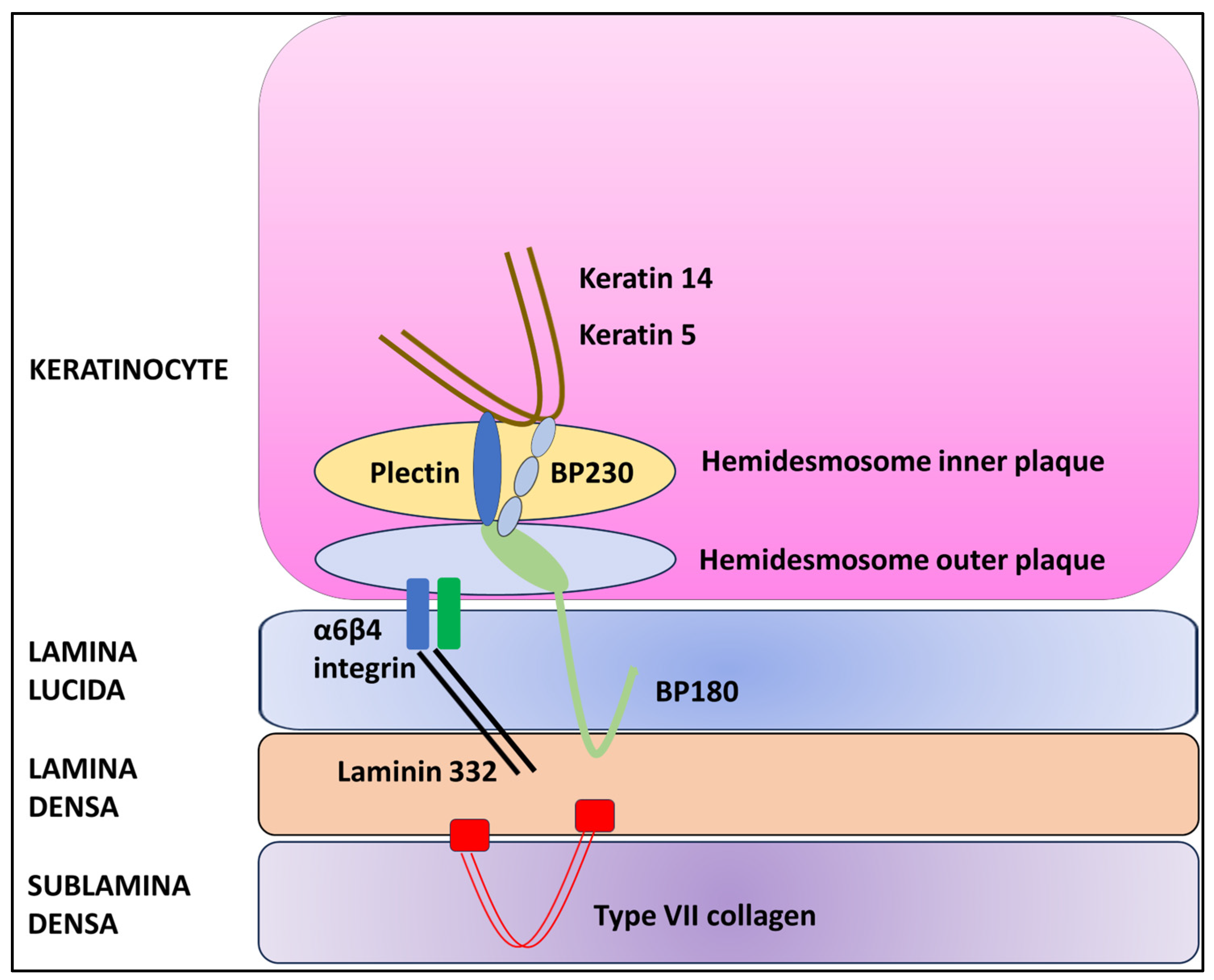
Figure 1. Schematic diagram of the dermal–epidermal junction.
In the presence of haploinsufficiency, the ratio of KRT5 to KRT14 is altered, resulting in a change in intermediate filament structure and impaired binding to desmosomes and hemidesmosomes and leading to the characteristic histopathologic alteration associated with GGD, i.e., acantholysis [7][14]. However, the precise pathogenetic mechanism that causes acantholysis is still unknown [7][17][18]. In addition, KRT5 plays a key role in epidermal differentiation and the uptake and degradation of melanosomes by keratinocytes [19]. In fact, mutations of the KRT5 gene have been associated with hyperplasia of epidermal ridges and the accumulation of melanin, resulting in the typical reticular hyperpigmentation [14][19].
Mutations in the KRT5 gene were identified in 21 out of 69 patients diagnosed with classical Galli–Galli disease [8][9][18][20][21][22][23][24]. The most common genetic alteration in the KRT5 gene is the c.418dupA missense mutation, which was found in 13 of the 21 patients with a KRT5 mutation [18][21][22][24]. Notably, this specific mutation has also been observed in DDD [5][7][18][21][22][24]. The c.418dupA mutation results in the insertion of an adenosine at position 418, causing a frameshift in the gene sequence [5][8].
Several de novo mutations have been identified in the KRT5 gene over the years in patients with classical GGD, most of which have not been linked to distinct phenotypes [24] An Arabian patient harbored a substitution of thymine for cytosine at position 2 (c.T2C), resulting in the destruction of the NlaIII endonuclease recognition site and a change in the codon of coding mRNA, which resulted in haploinsufficiency [20].
An Asian-American woman was identified as carrying a c.38dupG mutation, which led to the insertion of a guanine at position 38 in exon 1, subsequently causing a premature stop codon and resulting in KRT5 haploinsufficiency [8].
A Caucasian patient had the c.14C > A mutation, which resulted in the substitution of cytosine for adenosine [21].
A Caucasian patient with a segmental phenotype was found to carry a c.476C4T mutation. This mutation caused a substitution of leucine for proline at position 159, resulting in the production of a nonfunctional protein [23].
An Indian family was found to carry a mutation c.10C > T, in which cysteine was replaced by threonine at position 10 [9]. In addition to the classic symptoms, this family exhibited hyperpigmentation on the extremities and face. Furthermore, multiple hypopigmented macules were symmetrically distributed over the trunk.
3. POGLUT1
With the development of whole-exome sequencing (WES), mutations in POGLUT1 (encoding protein O-glucosyltransferase 1) have been shown to underlie some cases of GGD/DDD [13][25].
The POGLUT1 gene, which is located on chromosome 3q13.33, encodes a protein that plays a crucial role in posttranslational modifications within the Notch pathway, transferring glucose molecules to serine residues of epidermal growth factor-like (EGF-like) domains within the endoplasmic reticulum [13][26][27]. The addition of the O-glucose residues by POGLUT1 plays a crucial role in the folding of Notch pathway receptor proteins within the endoplasmic reticulum, which is essential for their transport to the cell surface (Figure 2) [26][27][28][29].
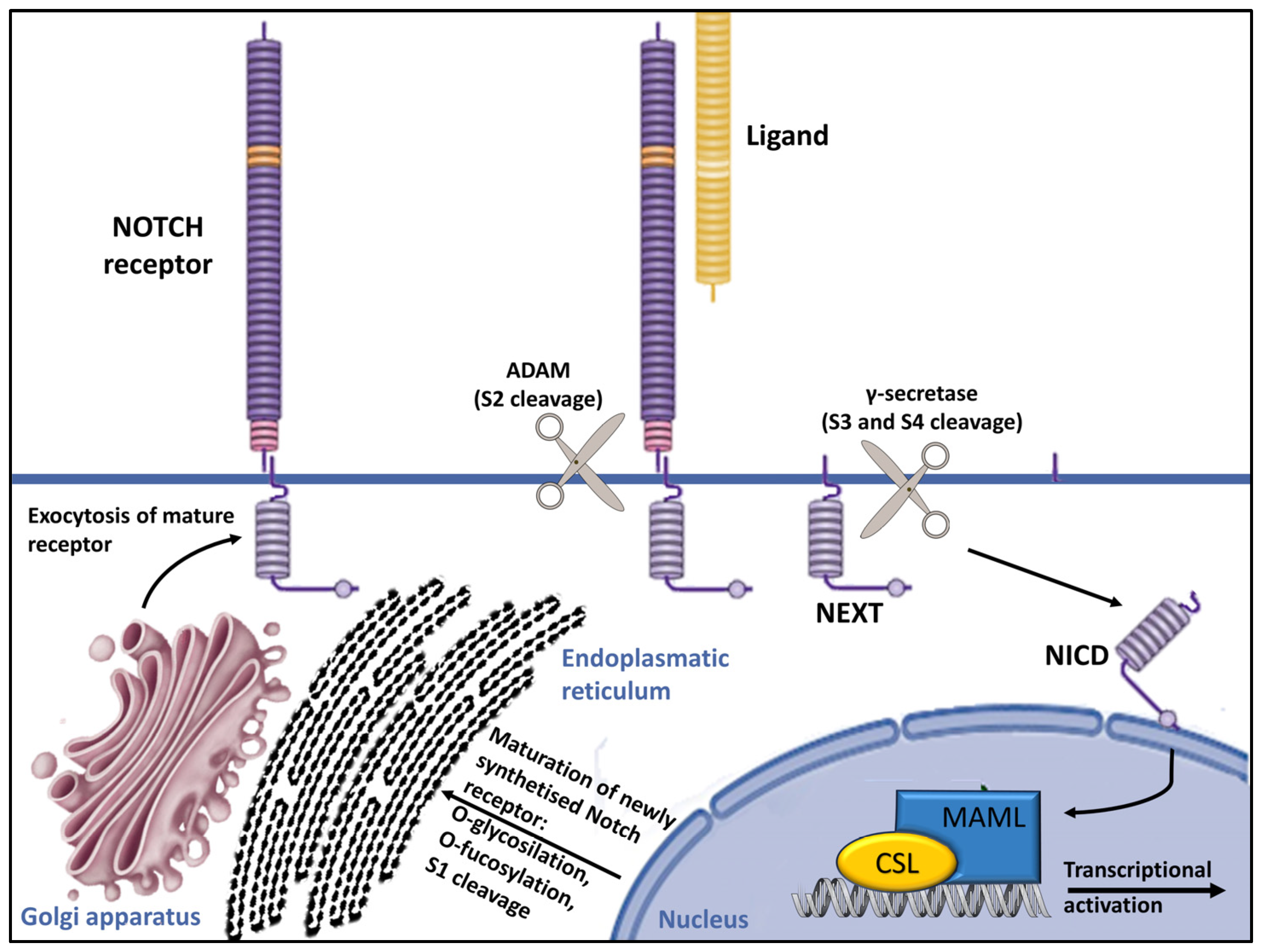
Figure 2. Overview of Notch signaling. Notch receptors undergo initial translation in the endoplasmic reticulum and are then trafficked to the Golgi apparatus. During trafficking, O-linked and N-linked glycosylations occur. Then, in the Golgi apparatus, Notch receptors are cleaved into heterodimers (S1 cleavage) and transported to the cell membrane. Upon reaching the cell surface, the receptor is activated through ligand binding. This ligand–receptor interaction triggers transendocytosis in the neighboring cell, inducing a conformational alteration in the Notch receptor and exposing the S2 site. This site is cleaved by ADAM metalloproteases (S2 cleavage), generating a membrane-anchored Notch extracellular truncation (NEXT). The S2 cleavage exposes S3 and S4 sites, facilitating further proteolytic cleavage by the γ-secretase complex (S3/S4 cleavage). This results in the liberation of the notch intracellular domain (NICD), which translocates to the nucleus. There, it binds to CBF1/RBP-Jκ/Su(H)/Lag-1 (CSL), which is known as RBP-Jκ in vertebrates. In its basal state, CSL functions as a transcriptional repressor by associating with corepressor (Co-R) proteins. NICD binding converts it into a transcriptional activator by forming a ternary complex with mastermind-like (MAML) proteins, thus initiating the transcription of downstream target genes [29].
The Notch pathway comprises four receptors and five ligands, and it is responsible for regulating skin homeostasis. It affects the proliferation and differentiation of melanocytes and keratinocytes, as well as their interaction. The alteration of this pathway leads to keratinocyte differentiation abnormalities, as evidenced by the increased expression of KRT5 and KRT14 in immunohistochemistry, as well as disturbance in skin pigmentation, as demonstrated by the abnormal migration of melanoblasts and melanocytes to ectopic sites in animal models [13].
In 2014, Basmanav et al. discovered various mutations in the POGLUT1 gene in individuals diagnosed with DDD who exhibited a disseminated pattern of brownish macular and lentiginous lesions on the extremities, trunk, and neck, rather than the typical domination of flexural folds commonly seen in classical DDD [13]. Notably, six of these cases had previously been classified as atypical GGD in an earlier study by one of the Authors [21]. They identified two nonsense mutations (c.11G > A and c.652C > T) and a splice-site mutation (c.798-2A > C) [13]. These mutations led to truncated proteins with compromised stability, subsequently reducing the glycosylation of receptor proteins in the Notch pathway [13].
The observed reduction in glycosylation has been demonstrated in animal models, revealing abnormalities in keratinocyte and melanocyte development and differentiation [13]. The POGLUT1 gene significance in cell differentiation is further supported by immunohistochemical analysis, which shows increased expression in the upper layers (spinous and granular) and a decrease in expression by about 50% in affected individuals. This is consistent with heterozygous mutations leading to a loss of function [13][25][27][30].
Histologically, the skin lesions of individuals with POGLUT1 mutations have showed a digitiform epidermal ridge hyperplasia with pronounced hyperpigmentation at the tips of the rete ridges, focal hypergranulosis, and, notably, some small horn cysts and minor acantholysis.
In recent years, three additional cases of GGD with POGLUT1 mutation were identified: one with a c.1013delTinsAAC [30], one with c.652 > T [4], and one with c.798-2A > C [31].
4. A Possible Role of PSENEN?
The presenilin enhancer, gamma-secretase subunit (PENSEN) gene plays a key role in Notch signaling activation, providing instructions for a protein called gamma-secretase subunit PEN-2 (shortened to PEN-2) [32]. It is a transmembrane protease composed of four essential protein subunits (the γ-secretase complex) located in the cell membrane, where it cleaves many different transmembrane proteins. This cleavage is an important step in several chemical signaling pathways that transmit signals from outside the cell into the nucleus, including Notch signaling. (Figure 2) [29][32][33][34][35].
The PSENEN gene, which has been extensively studied in the context of neurodegeneration, has been linked to over 300 mutations primarily associated with early-onset Alzheimer’s disease [32]. Recently, mutations in PSENEN have also been discovered in patients with familial hidradenitis suppurativa (HS) [36]. These mutations result in presenilin dysfunction and impaired cleavage activity, particularly affecting substrates like Notch receptors [37][38].
A cooccurrence of HS and DDD was first mentioned in 1990 [39] and subsequently confirmed in numerous case reports [33]. Pavlovsky et al. described four more individuals with HS–DDD [33] Importantly, the study also identified a founder mutation in the PSENEN gene [33].
Del Mar et al. reported the first case of GGD associated with HS but did not conduct any genetic assays to identify mutations that had been previously reported in such conditions [34]. Although the precise pathogenic mechanism linking GGD and DDD with HS remains unclear, current evidence suggests that mutations in PSENEN may be involved, similarly to alterations in POGLUT1, affecting the Notch signaling pathway [33][38][40][41].
References
- Gilchrist, H.; Jackson, S.; Morse, L.; Nesbitt, L.T. Galli-Galli disease: A case report with review of the literature. J. Am. Acad. Dermatol. 2008, 58, 299–302.
- Braun-Falco, M.; Volgger, W.; Borelli, S.; Ring, J.; Disch, R. Galli-Galli disease: An unrecognized entity or an acantholytic variant of Dowling-Degos disease? J. Am. Acad. Dermatol. 2001, 45, 760–763.
- Bardach, H.; Gebhart, W.; Luger, T. Genodermatose bei einem Brüderpaar: Morbus Dowling-Degos, Grover, Darier, Hailey-Hailey oder Galli-Galli? . Hautarzt 1982, 33, 378–383.
- Rundle, C.W.; Ophaug, S.; Simpson, E.L. Acitretin therapy for Galli-Galli disease. JAAD Case Rep. 2020, 6, 457–461.
- Yang, A.; Cheung, K.; Kossard, S.; Murrell, D.F. Atypical Disseminated Variant of Galli–Galli Disease: A Review of the Literature. Am. J. Dermatopathol. 2020, 42, 484–490.
- Deenen, N.J.; Damman, J.; Nijsten, T.E.C. Galli–Galli disease successfully treated with alitretinoin. J. Eur. Acad. Dermatol. Venereol. 2019, 33, e232–e233.
- Hanneken, S.; Rütten, A.; Pasternack, S.M.; Eigelshoven, S.; El Shabrawi-Caelen, L.; Wenzel, J.; Braun-Falco, M.; Ruzicka, T.; Nöthen, M.M.; Kruse, R.; et al. Systematic mutation screening of KRT5 supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. Br. J. Dermatol. 2010, 163, 197–200.
- Reisenauer, A.K.; Wordingham, S.V.; York, J.; Kokkonen, E.W.J.; McLean, W.H.I.; Wilson, N.J.; Smith, F.J.D. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br. J. Dermatol. 2014, 170, 1362–1365.
- Verma, S.; Pasternack, S.M.; Rutten, A.; Ruzicka, T.; Betz, R.C.; Hanneken, S. The first report of krt5 mutation underlying acantholytic dowling-degos disease with mottled hypopigmentation in an Indian family. Indian J. Dermatol. 2014, 59, 476–480.
- Stephan, C.; Kurban, M.; Abbas, O. Dowling-Degos disease: A review. Int. J. Dermatol. 2021, 60, 944–950.
- Kim, Y.C.; Davis, M.D.P.; Schanbacher, C.F.; Su, W.P.D. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): A clinical and histopathologic study of 6 cases. J. Am. Acad. Dermatol. 1999, 40, 462–467.
- Batycka-Baran, A.; Baran, W.; Hryncewicz-Gwozdz, A.; Burgdorf, W. Dowling-degos disease: Case report and review of the literature. Dermatology 2010, 220, 254–258.
- Basmanav, F.B.; Oprisoreanu, A.M.; Pasternack, S.M.; Thiele, H.; Fritz, G.; Wenzel, J.; Größer, L.; Wehner, M.; Wolf, S.; Fagerberg, C.; et al. Mutations in POGLUT1, encoding protein o-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos Disease. Am. J. Hum. Genet. 2014, 94, 135–143.
- Betz, R.C.; Planko, L.; Eigelshoven, S.; Hanneken, S.; Pasternack, S.M.; Büssow, H.; Van Den Bogaert, K.; Wenzel, J.; Braun-Falco, M.; Rütten, A.; et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am. J. Hum. Genet. 2006, 78, 510–519.
- Bouameur, J.E.; Favre, B.; Fontao, L.; Lingasamy, P.; Begré, N.; Borradori, L. Interaction of plectin with keratins 5 and 14: Dependence on several plectin domains and keratin quaternary structure. J. Investig. Dermatol. 2014, 134, 2776–2783.
- Intong, L.R.A.; Murrell, D.F. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin. Dermatol. 2012, 30, 70–77.
- Müller, C.S.; Tremezaygues, L.; Pföhler, C.; Vogt, T. The spectrum of reticulate pigment disorders of the skin revisited. Eur. J. Dermatol. 2012, 22, 596–604.
- Lőrincz, K.; Medvecz, M.; Kiss, N.; Glász-Bóna, A.; Hársing, J.; Lepesi-Benkő, R.; Hatvani, Z.; Mazán, M.; Kárpáti, S.; Wikonkál, N. Confirmation of the role of a KRT5 mutation and successful management of skin lesions in a patient with Galli–Galli disease. Clin. Exp. Dermatol. 2018, 43, 972–974.
- El Shabrawi-Caelen, L.; Rütten, A.; Kerl, H. The expanding spectrum of Galli-Galli disease. J. Am. Acad. Dermatol. 2007, 56, S86–S91.
- Sprecher, E.; Indelman, M.; Khamaysi, Z.; Lugassy, J.; Petronius, D.; Bergman, R. Galli-Galli disease is an acantholytic variant of Dowling-Degos disease. Br. J. Dermatol. 2007, 156, 572–574.
- Hanneken, S.; Rütten, A.; Eigelshoven, S.; Braun-Falco, M.; Pasternack, S.M.; Ruzicka, T.; Nöthen, M.M.; Betz, R.C.; Kruse, R. Klinische und histopathologische Untersuchung anhand einer Fallserie von 18 Patienten . Hautarzt 2011, 62, 842–851.
- Voth, H.; Landsberg, J.; Reinhard, G.; Refke, M.; Betz, R.C.; Bieber, T.; Wenzel, J. Efficacy of Ablative Laser Treatment in Galli-Galli Disease. Arch. Dermatol. 2011, 147, 317.
- Arnold, A.W.; Kiritsi, D.; Happle, R.; Kohlhase, J.; Hausser, I.; Bruckner-Tuderman, L.; Has, C.; Itin, P.H. Type 1 segmental galli-galli disease resulting from a previously unreported keratin 5 mutation. J. Investig. Dermatol. 2012, 132, 2100–2103.
- Schmieder, A.; Pasternack, S.M.; Krahl, D.; Betz, R.C.; Leverkus, M. Galli-Galli disease is an acantholytic variant of Dowling-Degos disease: Additional genetic evidence in a German family. J. Am. Acad. Dermatol. 2012, 66, e250–e251.
- Wilson, N.J.; Cole, C.; Kroboth, K.; Hunter, W.N.; Mann, J.A.; McLean, W.H.I.; Kernland Lang, K.; Beltraminelli, H.; Sabroe, R.A.; Tiffin, N.; et al. Mutations in POGLUT1 in Galli–Galli/Dowling–Degos disease. Br. J. Dermatol. 2017, 176, 270–274.
- Mehboob, M.Z.; Lang, M. Structure, function, and pathology of protein O-glucosyltransferases. Cell Death Dis. 2021, 12, 71.
- Yu, H.; Takeuchi, H. Protein O-glucosylation: Another essential role of glucose in biology. Curr. Opin. Struct. Biol. 2019, 56, 64–71.
- Li, Z.; Han, K.; Pak, J.E.; Satkunarajah, M.; Zhou, D.; Rini, J.M. Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol. 2017, 13, 757–763.
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95.
- Kono, M.; Sawada, M.; Nakazawa, Y.; Ogi, T.; Muro, Y.; Akiyama, M. A Japanese case of galli-galli disease due to a previously unreported POGLUT1 mutation. Acta Derm. Venereol. 2019, 99, 458–459.
- Seitz, A.T.; Sterz, H.; Strehlow, V.; Nagel, S.; Dumann, K.; Grunewald, S.; Simon, J.C.; Kunz, M. Full ablative versus fractional ablative laser therapy for Dowling–Degos disease. Lasers Surg. Med. 2018, 51, 321–324.
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene. Int. J. Mol. Sci. 2022, 23, 10970.
- Pavlovsky, M.; Sarig, O.; Eskin-Schwartz, M.; Malchin, N.; Bochner, R.; Mohamad, J.; Gat, A.; Peled, A.; Hafner, A.; Sprecher, E. A phenotype combining hidradenitis suppurativa with Dowling–Degos disease caused by a founder mutation in PSENEN. Br. J. Dermatol. 2018, 178, 502–508.
- Del Mar, M.; González, M.; Sayed, C.; Phadke, P. Hidradenitis Suppurativa Associated with Galli-Galli Disease: Extending the link with Dowling-Degos Disease. JCAD J. Clin. Aesthetic Dermatol. 2020, 13, 46–47.
- Wang, Z.; Yan, Y.; Wang, B. γ-Secretase Genetics of Hidradenitis Suppurativa: A Systematic Literature Review. Genet. Artic. Dermatol. 2021, 237, 698–704.
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. γ-Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065.
- Zhou, C.; Wen, G.D.; Soe, L.M.; Xu, H.J.; Du, J.; Zhang, J.Z. Novel mutations in PSENEN gene in two Chinese acne inversa families manifested as familial multiple comedones and dowling-degos disease. Chin. Med. J. 2016, 129, 2834–2839.
- Ralser, D.J.; Basmanav, F.B.; Tafazzoli, A.; Wititsuwannakul, J.; Delker, S.; Danda, S.; Thiele, H.; Wolf, S.; Busch, M.; Pulimood, S.A.; et al. Mutations in γ-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J. Clin. Investig. 2017, 127, 1485–1490.
- Weber, L.A.; Kantor, G.R.; Bergfeld, W.F. Reticulate Pigmented Anomaly of the Flexure (DDD): A case report associated with Hidradenitis Suppurativa and Squamous Cell Carcinoma. Cutis 1990, 45, 446–450.
- Pink, A.E.; Simpson, M.A.; Brice, G.W.; Smith, C.H.; Desai, N.; Mortimer, P.S.; Barker, J.N.W.N.; Trembath, R.C. PSENEN and NCSTN mutations in familial Hidradenitis suppurativa (Acne inversa). J. Investig. Dermatol. 2011, 131, 1568–1570.
- Peter, D.C.V.; Smith, F.J.D.; Wilson, N.J.; Danda, S. PSENEN Mutation in Coexistent Hidradenitis Suppurativa and Dowling-Degos Disease. Indian Dermatol. Online J. 2020, 12, 147–149.
More
Information
Subjects:
Dermatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
693
Revisions:
2 times
(View History)
Update Date:
26 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No