Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aleksey Shchulkin | -- | 3523 | 2024-02-23 12:20:17 | | | |
| 2 | Camila Xu | Meta information modification | 3523 | 2024-02-26 01:33:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shchulkin, A.V.; Abalenikhina, Y.V.; Kosmachevskaya, O.V.; Topunov, A.F.; Yakusheva, E.N. Regulation of P-glycoprotein. Encyclopedia. Available online: https://encyclopedia.pub/entry/55389 (accessed on 11 June 2026).
Shchulkin AV, Abalenikhina YV, Kosmachevskaya OV, Topunov AF, Yakusheva EN. Regulation of P-glycoprotein. Encyclopedia. Available at: https://encyclopedia.pub/entry/55389. Accessed June 11, 2026.
Shchulkin, Aleksey V., Yulia V. Abalenikhina, Olga V. Kosmachevskaya, Alexey F. Topunov, Elena N. Yakusheva. "Regulation of P-glycoprotein" Encyclopedia, https://encyclopedia.pub/entry/55389 (accessed June 11, 2026).
Shchulkin, A.V., Abalenikhina, Y.V., Kosmachevskaya, O.V., Topunov, A.F., & Yakusheva, E.N. (2024, February 23). Regulation of P-glycoprotein. In Encyclopedia. https://encyclopedia.pub/entry/55389
Shchulkin, Aleksey V., et al. "Regulation of P-glycoprotein." Encyclopedia. Web. 23 February, 2024.
Copy Citation
P-glycoprotein (Pgp, ABCB1 protein, MDR1) is a transporter protein, the most studied representative of the ABC transporter superfamily. Pgp is an efflux protein: it ensures the outflow of molecules from cells into the extracellular space.
P-glycoprotein
oxidative stress
reactive oxygen species
Nrf2
Nf-kB
1. Introduction
P-glycoprotein (Pgp, ABCB1 protein, MDR1) is a transporter protein, the most studied representative of the ABC transporter superfamily [1]. Pgp is an efflux protein: it ensures the outflow of molecules from cells into the extracellular space. The main function of Pgp is excreting xeno- and endobiotics into the extracellular space, biological fluids (blood, bile and urine) and intestinal lumen. Pgp plays an important role in pharmacokinetics—absorption, distribution, and excretion of drugs, which are its substrates; is involved in the transport of endogenous molecules (steroid and thyroid hormones); and contributes to the resistance of tumor cells to chemotherapy [2][3][4][5]. However, the hypothesis that the sole function of Pgp is to remove xenobiotics from the cell does not explain the high level of the transporter protein in the adrenal glands [6] or its apical localization in the epithelial cells of the vascular plexus [7]. Pgp is also able to block the development of apoptosis in tumor cells by modulating the activity of key enzymes for their programmed death [8][9]. Thus, Pgp plays a key role not only in the pharmacokinetics of drugs, being its substrates, but also in physiological and pathological processes.
The activity of Pgp can vary significantly under the influence of the external and internal environmental factors, such as genetic characteristics of the body, oxygen concentration in blood, acid–base balance, the use of a number of drugs, etc. [10]. According to modern concepts, Pgp activity can change as a result of the following main processes: modulation of the expression of the multidrug resistance gene (MDR1, multidrug resistance gene) and the activity of the synthesized protein [11][12]. At the same time, the activity of Pgp can both decrease (inhibition) and increase (induction) [13][14]. That is why the regulation mechanisms of the transporter protein are currently being actively studied.
Studies from the early 1960s to the 1980s showed that the increased production of reactive oxygen species (ROS) plays an important role in the pathogenesis of the most common human diseases (pathology of the cardiovascular, respiratory, endocrine systems and cancers) [15][16][17]. Mainly lipid and protein molecules are subject to free radical oxidation in cells [18][19][20][21]. The free radical oxidation of biomolecules leads to oxidative stress (OS), characterized by highly toxic oxidation products accumulated in blood and tissues [20]. Oxidative modification of both lipids and proteins changes the viscosity, elasticity and fluidity of membranes, which significantly affects the cell–cell interaction, mitosis and endocytosis [22]. Therefore, a number of researchers consider the cell membrane as a biosensor of the OS [23].
Pgp is localized not only in plasma membranes but also in the membranes of intracellular compartments (endoplasmic reticulum, Golgi apparatus, lysosomes and mitochondria), and the role of the transporter in these compartments continues to be studied [24][25]. Free radical oxidation of membrane lipids in different compartments may disrupt the activity of Pgp. That is why the etiology of OS development (exogenous or endogenous formation of ROS) is important in studying the regulation of Pgp activity and expression. In addition, the peroxidation products, formed as a result of OS, may directly or indirectly affect the functioning of Pgp or its substrates. The functioning of the transporter protein is of great practical importance due to the role of Pgp in the pharmacokinetics of a wide range of drugs, as well as its contribution to the development of pharmacotherapy-resistant diseases (epilepsy and tumors). In this regard, studying OS’s effect on the activity of Pgp will significantly improve the effectiveness and safety of the ongoing therapy by predicting pharmacokinetics and adverse drug reactions.
2. Pgp: Localization, Structure, Inhibitors and Substrates and Mechanisms of Regulation
2.1. Brief History and Overview of Pgp
Pgp was first isolated in 1976 by Juliano and Ling from the plasma membrane of Chinese hamster ovarian cells, selected for colchicine resistance and demonstrating cross-resistance to a wide range of amphiphilic substances [26]. The protein was named P-glycoprotein (P—permeability) because it was believed to be involved in the development of drug resistance via reducing the permeability of the cell membrane [27]. Ten years later, the genes responsible for multidrug resistance (MDR genes) were cloned in humans, mice and hamsters [28][29][30], and Pgp was shown to be the protein product of the MDR1 gene [31]. Subsequently, Fojo et al. (1987) demonstrated that the gene encoding Pgp—MDR1 in humans—is highly expressed in the adrenal glands and kidneys, at an average level in the lungs, liver, jejunum, colon and rectum (samples were obtained by paracentesis) [32].
Thiebaut et al. (1987) immunohistochemically studied Pgp localization in human tissues obtained during autopsy or surgical operations [33]. They showed that Pgp is localized in the liver, mainly on the biliary surface of hepatocytes and on the apical surface of epithelial cells in small biliary ducts. In the pancreas, Pgp was found on the apical surface of epithelial cells of small ducts, but not of large ones. In kidneys, it was found on the apical surface of the epithelial cells of the proximal renal tubules. Colon and jejunum showed high levels of Pgp on the apical surface of epithelial cells. The adrenal glands were characterized by high levels of Pgp on the cell surface in the medulla and cortex [33]. In 1994, Schinkel et al. revealed the expression of mdr1a in the endothelial cells of the capillaries in the blood–brain barrier [34]. Its expression was also found in cells on the luminal surface of the endometrium of the pregnant uterus, and also in placental trophoblasts [35].
Initially, Pgp was supposed to protect tumor cells from the effect of cytostatics by removing them from the cells [36]. However, it was later shown that Pgp performs an important function of transporting endogenous substances and also protects cells from the effects of xenobiotics, providing their efflux into the extracellular space and biological fluids (blood, bile and urine) [37][38]. In the blood–brain barrier, Pgp protects the brain from exposure to toxic substances. In mice with Pgp gene knockout, the concentration of xenobiotic in the brain increased 20–80 times, while the levels of xenobiotics in other organs, such as liver, kidneys and intestines, increased only 2–4 times [34]. Compared to the wild-type mice, mice with Pgp gene knockout require a lower dose of the drug pilocarpine to induce seizures [39]. In mammalian kidneys, Pgp ensures the excretion of drugs, their conjugates and metabolites into the lumen of the proximal renal tubules [40]. For example, in the canine kidney, tubule cells of Madin Darby (MDCK) transfected with the ABCB1 gene encoding Pgp, the basolateral to apical outflow of the oncological drug gefitinib, was significantly increased compared to the corresponding control cells. In the presence of Pgp inhibitor LY335979, the outflow of gefitinib in cells transfected with the MDR1 gene decreased to the same level as in the control cells [41].
In hepatocytes, Pgp is expressed on the apical (biliary) surface, ensuring the excretion of xenobiotics into bile. Furthermore, Pgp inhibition can also affect bile excretion [42][43][44][45]. In enterocytes, Pgp is expressed on the apical surface and prevents the uptake of substrates into the systemic blood flow [45][46][47].
Pgp transports a wide range of structurally and functionally different cytotoxic compounds out of the cell. Overexpression of Pgp in cancer cells, compared to normal cells, is the main cause of multidrug resistance (MDR). Therefore, the inclusion of Pgp inhibitors in drugs can reduce Pgp activity in cancer cells [48] and thereby increase therapeutic efficacy [49]. Pgp is an essential component in the drug distribution process, contributing to drug–drug interactions [50] (Figure 1).
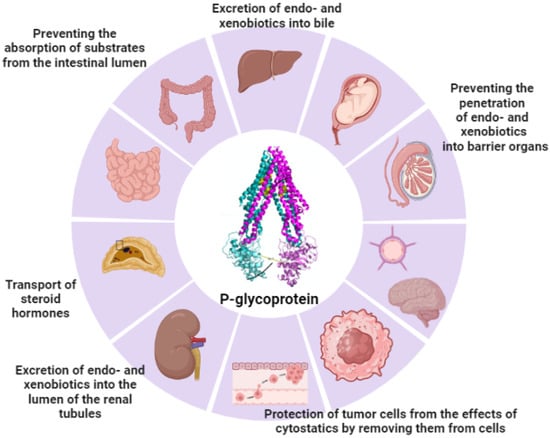
Figure 1. Localization and functions of Pgp (the figure was made using app.biorender.com, accessed on 2 October 2023).
2.2. Structure of Pgp and Its Substrates and Inhibitors
Pgp is an N-glycosylated transmembrane glycoprotein that weighs 170 kDa. It consists of 1280 amino acid residues in humans and of 1276 amino acid residues in mice (bears 87% sequence identity to human Pgp) grouped into two homologous halves, which are connected to each other by a mobile linker polypeptide (Supplementary Materials) [51][52].
Each part consists of an NH2-terminal transmembrane domain (TMD) containing six transmembrane (TM) segments (the so-called hydrophobic segments or α-helices) interconnected by several intracellular and extracellular loops (hydrophilic loops), as well as a hydrophilic (intracellular) domain containing ATP-binding sites (NBDs), which are placed inside the plasma membrane. Analyzing the secondary structure of Pgp showed that the transporter protein consists of 32–43% α-helices, 16–26% β-folded layers (β-structures, β-sheets), 15–29% β-bends and 13–26% irregular secondary structures (Figure 2) [53][54][55].
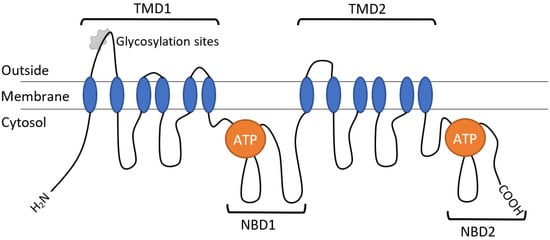
Figure 2. Schematic representation of the structure of the human Pgp transporter protein.
ATP energy is necessary for the Pgp operation and for the stabilization of its external conformation [56][57].
Currently, the tertiary structure of a multidrug-resistant protein remains unclear, mainly due to the difficulties associated with protein crystallization. The tertiary structure of Pgp is known to be highly flexible, providing for three-dimensional reorientation. This characteristic of Pgp further facilitates its interaction with a wide range of substrates [58][59].
To enter the hydrophobic binding pocket of the Pgp and pass through the membrane, the molecule must be lipophilic. Free energy calculating showed that hydrophobic fragments mainly contribute to ligand binding. Many studies suggest that the hydrophobicity of the molecule is crucial to the development of transporter inhibitors. On the contrary, the hydrophilicity of the molecule may interfere with Pgp efflux [60].
Nonpolar, linear, hydrophobic and aromatic compounds with different molecular weights, ranging from 250 to 4000 Da, were identified as Pgp substrates [61][62]. The Pgp substrates are sphingolipids; platelet activation factor; short-chain phospholipids; cytokines; and lipophilic drugs—antitumor drugs, immunosuppressants, steroid and thyroid hormones, antibiotics, HIV proteinase inhibitors, cardiac glycosides, anticoagulants, etc. [10][50].
Using computational methods and programming, combined with structural design [63], the necessary characteristics of inhibitors that are potentially suitable for clinical use were determined:
- (a)
-
A high log p-value (determined by the lipid and water distribution coefficient for the drug). This parameter for the compound must be at least 2.92 or higher, which is necessary for the formation of a hydrophobic/van der Waals interaction with the Pgp binding site;
- (b)
-
Significant molecular weight—A molecule must have 18 or more atoms to cover more than one Pgp binding site;
- (c)
-
The energy of the highest occupied molecular orbital (highest occupied molecular orbital, HOMO) should have a higher value to ensure the nucleophilic interaction of the molecule with Pgp;
- (d)
-
At least one tertiary nitrogen atom since the tertiary amine forms a cation at physiological pH and guarantees binding by ionic interaction.
Many studies report on the aromatic rings; molecular weight; cationic charge, such as protonated amine; and H-bond donor/acceptor factors. In general, inhibitors have a higher log p-value than substrates. These inhibitors act primarily as H-bond donors rather than as H-bond acceptors. Functional groups, including arene, alkyl, carbonyl, ether and nitrogen ones, are necessary for strong interactions between the protein and the inhibitor, thereby affecting the effectiveness and pharmacodynamic aspect of the interactions. Studies on the dependence of structure and activity have shown that lipophilicity and the value of log ligands (substrate and inhibitor) are important parameters affecting pharmacokinetic aspects [64][65].
Pgp inhibition can also occur as a result of changes in its structure. For example, flavonoids [66], xanthones [67] and synthetic taxoids (derivatives of the antitumor agent taxol) cause conformational changes in Pgp, and such changes, in turn, disrupt the outflow function due to the formation of H-bonds and ATP hydrolysis [68].
2.3. Mechanisms of Regulation of Pgp
The following mechanisms of Pgp regulation are distinguished.
- (1)
-
Mechanisms of the MDR1 gene expression regulation.
Pgp in humans is encoded by the MDR1 gene, while in mice and rats, it is encoded by the mdr1a (also called mdr3) and mdr1b (also called mdr1) genes, and the homologous halves of Pgp are the product of duplication of the MDR1 gene [34][69][70]. It was previously assumed that, in humans, Pgp can also be encoded by the MDR3 gene (sometimes referred to as MDR2) and the mdr2 gene in mice. However, these genes were later shown not to be involved in multidrug resistance and to encode phospholipid flippase expressed in the biliary membranes of hepatocytes. They were revealed to play an important role in the secretion of phosphatidylcholine into bile. According to the international classification, this gene is designated as ABCB4 [69]. An increase in the expression of the MDR1 gene increases Pgp activity, and a decrease in its expression leads to its decrease. The MDR1 gene can be transcribed from two promoters—distal and proximal [70][71]. Proximal promoter regulates the expression of MDR1 in normal tissues, including the liver, kidneys and adrenal glands, while the distal promoter triggers the expression of MDR1 in colchicine-selected (selected) cells [32][72], mononuclear cells of patients with acute lymphoblastic leukemia who overexpress MDR1 [73], and cells of the primary breast tumors [74]. The effect of the nucleotide sequence of the MDR1 gene promoter on its transcription and expression was studied using luciferase as a reporter of this gene. The expression of the MDR1 gene can be influenced by both environmental factors and chemicals [75].
- (2)
-
Polymorphism of the MDR1 gene.
Screening of the MDR1 gene revealed about 50 substitutions of one nucleotide for another, the so–called polymorphisms of one nucleotide—SNP (single-nucleotide polymorphisms) [76][77]. Most polymorphisms of the MDR1 gene do not significantly affect the expression and functional activity of Pgp [78][79]. Recently, analyzing whole haplotypes rather than the isolated polymorphisms of a single nucleotide was proposed [80].
- (3)
-
Increasing the dose of the gene—amplification of a section of the genome containing the MDR1 gene.
Kitada et al. revealed an increase in Pgp activity due to an increase in the dose (amplification) of the MDR1 gene. When treating the PTX250 lung cancer cell line with paclitaxel, the number of MDR1 gene copies was shown to increase 11 times, while the amplicon size was 2.7 megabytes [81]. It was also found that lung cancer cell sublines No. 15-80-1 and No. 15-80-6, which are obtained when treating the NCI-H460 cell line with increasing doses of the inducer Pgp paclitaxel (from 50 to 800 nM), are characterized by amplification of the MDR1 gene with a different number of copies. However, a common pattern of amplification is accompanied by an increase in Pgp activity [81].
- (4)
-
Stabilization of the mRNA of the MDR1 gene.
The Pgp activity may increase in the background of the stabilization of the mRNA of the MDR1 gene. For example, in myelogenous leukemic K562 cells, the level of MDR1 gene mRNA was found to be independent of the transcriptional activity, but it was regulated at posttranscriptional stages: mRNA stability and translational regulation. In particular, the short mRNA life of the MDR1 gene of native cells (1 h) increased to 12–16 h after short-term exposure to Pgp inducers—colchicine and doxorubicin [82]. In another study, it was shown that, during the treatment of T-cell leukemia cell culture with cytostatic cytarabine for 36 h, the half-life of the mRNA of the MDR1 gene increased from 30 min to 6 h [82]. At the same time, an increase in the stability of Pgp mRNA was accompanied by an increase in the amount of Pgp protein only with the long-term selection of cytostatic-resistant cells, and the short-term stabilization of mRNA did not lead to an increase in Pgp synthesis and activity. These results indicate that cell resistance to cytostatics is associated with two processes: stabilizing the mRNA of the MDR1 gene; and the overcoming of translational block, which is necessary to start Pgp synthesis [83].
- (5)
-
The effect of microRNAs on Pgp expression.
Several microRNAs were described as direct regulators of Pgp expression [84]. For example, microRNA-451 was shown to regulate Pgp expression through direct interaction with the 3′-untranslated region (3′-UTR) of MDR1 mRNA. An increase in the content of microRNA-451 in the doxorubicin-resistant breast cancer cell line (MCF-7-DOX) led to a decrease in Pgp expression and, more importantly, to an increase in cellular sensitivity to doxorubicin [85]. An inverse correlation between the levels of Pgp and microRNA-331-5p was found. It was additionally shown that overexpression of microRNA-331-5p increases the sensitivity of K562-DOX cells to doxorubicin by inhibiting Pgp expression [86]. Impaired microRNA-298 formation (due to low expression of the Dicer enzyme) was associated with modulating Pgp expression. MicroRNA-298 was shown to bind directly to the 3′-untranslated region (3′-UTR) of the mRNA of the MDR1 gene. The decreased level of this microRNA was accompanied by an increase in Pgp expression and the development of doxorubicin resistance in breast cancer cells (MDA-MB-231) [87]. In another study on a human colon carcinoma cell line (Caco-2), a decrease in the content of microRNA-145 increased the expression and activity of Pgp, but not the amount of mRNA of the MDR1 gene, indicating that, in this cell line, microRNA-145 regulates Pgp through translational mRNA repression [88]. Several microRNAs were described as “indirect regulators” of Pgp. These microRNAs interacting with mRNAs encoding intermediate proteins or transcription factors indirectly participate in activating the MDR1 gene [84]. For example, microRNA-let-7 was shown to regulate MDR1 in ovarian cancer cells by suppressing IMP-1, an RNA-binding protein, which destabilizes MDR1 mRNA and, as a consequence, decreases Pgp expression [89]. This indicates the possibility of indirect influence of these microRNAs on the protein transporter [90].
- (6)
-
Pgp transmission between cells.
All cells, prokaryotes and eukaryotes, release extracellular vesicles as part of their normal physiology and during acquired abnormalities. Depending on the cell of origin, extracellular vesicles can contain many constituents of a cell, including DNA, RNA, lipids, metabolites, and cytosolic and cell-surface proteins [91]. The ability of the Adriamycin-resistant culture of bladder cells (BIU87) to transmit Pgp to sensitive cells by using microparticles of the plasma membrane (microparticles) was revealed. At the same time, different cell cultures were incubated for 48 h in media, separated by a membrane that prevented their direct contact. The presence of Pgp on the recipient cell membranes was determined via the Western blot method, and its functioning was confirmed by the active efflux of rhodamine-123, a substrate of the transporter protein [92]. In vitro and in vivo studies showed that the transfer of protein transporter can occur between cells of various origins: tumor and unchanged; and human and mouse. It was revealed that Pgp is transported via binding to microparticles of plasma membranes larger than 0.8 microns in size, while there is no cell fusion or formation of gap contacts between them [93]. Pgp transmission to sensitive tumor cells inhibits their growth and forms an unstable resistant state. It allows them to survive in an environment containing high doses of chemotherapeutic agents for a time, sufficient to acquire their own resistance due to the independent expression of Pgp [93].
- (7)
-
Changes in the activity of the synthesized transporter protein.
The activity of Pgp can change as a result of the direct interaction of the transporter protein molecule with molecules of endogenous and exogenous substances. Meanwhile, Pgp activity can both decrease (inhibition) and increase (induction) [94][95]. To date, three ways to inhibit the transporter protein activity have been described: competitive, noncompetitive and allosteric [96]. In competitive and noncompetitive inhibition, the inhibitor interacts with binding centers of substrates, and in the allosteric one, with the allosteric center. Data on these centers were summarized in papers by Martin et al. (2000) [97], Ferreira et al. (2013) [98] and Bocci et al. (2018) [99]. The authors identified three binding sites of substrates (transported substances) and one regulatory (allosteric, M-site) site that changes the activity of transporter protein. The three sites of substrate binding were named the D, R and H sites, according to the names of bound substrates—digoxin, rhodamine-123 and hext-33342, respectively. The competitive mechanism of Pgp activity inhibition assumes that the inhibitor binds to one, two or three sites and inactivates them. At the same time, if there are free binding sites, they can transport their substrates. For example, terfenadine is capable of inhibiting the transfer of Pgp hext-33342 and rhodamine-123, but not that of digoxin. Prazosin blocks the transport of digoxin and hext-33342, but not that of rhodamine-123. On the contrary, some other compounds, such as reserpine and loperamide, interfere with the transport of three Pgp substrates [99][100].
- (8)
-
Effect on ATP hydrolysis (ATPase activity).
Pgp is an ATP-dependent transporter protein, implicating ATP energy at the cornerstone of its functioning. In a study on cells overexpressing the protein, it was found that sodium azide—an inhibitor of metabolic processes—reduces its activity [101]. Some flavonoids (e.g., quercetin) also reduce Pgp activity by inhibiting ATPase activity of the protein transporter. Their advantage is that they do not compete with substrates for binding centers and inhibit the transporter protein at lower concentrations [96].
- (9)
-
Changes in plasma membrane characteristics.
Many surfactants, such as sodium dodecyl sulfate, Tween-20 and Span-80, change the characteristics of membrane lipids and, as a consequence, the fluidity (viscosity) of membranes, which, in turn, can inhibit Pgp activity [102]. A change in the microviscosity of the membrane can contribute to a change in the conformation of most transmembrane proteins. For example, the modification of the secondary and tertiary structure by surfactants was shown to decrease Pgp activity [96]. Recently, other substances inhibiting Pgp by increasing membrane fluidity were described, for example, a new synthetic derivative of rifampicin, 1,8-dibenzoylrifampicin, formed by introducing hydrophobic substituents into a positively charged rifampicin molecule. Although this substance has not yet been tested in vivo, it may be an interesting strategy for increasing the therapeutic activity of Pgp substrates [103].
Among all mechanisms of Pgp regulation, changes in the expression of the MDR1 gene and changes in the activity of the synthesized transporter protein are of paramount importance.
References
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290.
- Borst, P.; Schinkel, A.H. P-glycoprotein ABCB1: A major player in drug handling by mammals. J. Clin. Investig. 2013, 123, 4131–4133.
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178.
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98.
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123.
- Sugawara, I. Expression and functions of P-glycoprotein (mdr1 gene product) in normal and malignant tissues. Acta Pathol. Jpn. 1990, 40, 545–553.
- On, N.H.; Chen, F.; Hinton, M.; Miller, D.W. Assessment of P-glycoprotein activity in the Blood-Brain Barrier (BBB) using Near Infrared Fluorescence (NIRF) imaging techniques. Pharm. Res. 2011, 28, 2505–2515.
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into P-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170.
- Zu, Y.; Yang, Z.; Tang, S.; Han, Y.; Ma, J. Effects of P-glycoprotein and its inhibitors on apoptosis in K562 cells. Molecules 2014, 19, 13061–13075.
- Kukes, V.G.; Grachev, S.V.; Sychev, D.A.; Ramenskaya, G.V. Drug Metabolism. Scientific Foundations of Personalized Medicine: A Guide for Doctors; Geotar-Media: Moscow, Russia, 2008; p. 304.
- Yakusheva, E.N.; Titov, D.S. Structure and function of multidrug resistance protein 1. Biochemistry 2018, 83, 907–929.
- Iakusheva, E.N.; Chernykh, I.V.; Shchul’kin, A.V.; Popova, N.M. P-glycoprotein: Structure, physiological role and molecular mechanisms of modulation functional activity. Usp. Fiziol. Nauk. 2014, 45, 89–98.
- Sterz, K.; Möllmann, L.; Jacobs, A.; Baumert, D.; Wiese, M. Activators of P-glycoprotein: Structure-activity relationships and investigation of their mode of action. Chem. Med. Chem. 2009, 4, 1897–1911.
- Nguyen, T.-T.-L.; Duong, V.-A.; Maeng, H.-J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103.
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; p. 905.
- Sies, H. (Ed.) Oxidative Stress; Academic Press: London, UK, 1985; pp. 1–507.
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268.
- Vladimirov, Y.A. Free-radical oxidation of lipids and the physical properties of the lipid layer of biological membranes. Biophysics 1987, 32, 830–844.
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425.
- Gebicki, J.M.; Du, J.; Collins, J.; Tweeddale, H. Peroxidation of proteins and lipids in suspensions of liposomes, in blood serum, and in mouse myeloma cells. Acta Biochim. Pol. 2000, 47, 901–911.
- Bongarzone, E.R.; Pasquini, J.M.; Soto, E.F. Oxidative damage to proteins and lipids of CNS myelin produced by in vitro generated reactive oxygen species. J. Neurosci. Res. 1995, 41, 213–221.
- Tai, W.Y.; Yang, Y.C.; Lin, H.J.; Huang, C.P.; Cheng, Y.L.; Chen, M.F.; Yen, H.L.; Liau, L. Interplay between structure and fluidity of model lipid membranes under oxidative attack. J. Phys. Chem B 2010, 114, 15642–15649.
- Benderitter, M.; Vincent-Genod, L.; Pouget, J.P.; Voisin, P. The cell membrane as a biosensor of oxidative stress induced by radiation exposure: A multiparameter investigation. Radiat. Res. 2003, 159, 471–483.
- Fu, D.; Arias, I.M. Intracellular trafficking of P-glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464.
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167.
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutans. Biochem. Biophis. Acta 1976, 455, 155–162.
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009.
- Roninson, I.B.; Abelson, H.T.; Housman, D.E.; Howell, N.; Varshavsky, A. Amplification of specific DNA sequences correlates with multi-drug resistance in Chinese hamster cells. Nature 1984, 309, 626–628.
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542.
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased mdr1 expression can precede gene amplification. Science 1986, 232, 643–645.
- Ueda, K.; Cornwell, M.M.; Gottesman, M.M.; Pastan, I.; Roninson, I.B.; Ling, V.; Riordan, J.R. The mdr1 gene, responsible for multidrug-resistance, codes for Pglycoprotein. Biochem. Biophys. Res. Commun. 1986, 141, 956–962.
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269.
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738.
- Schinkel, A.H.; Smit, J.J.M.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; Vandeemter, L.; Mol, C.; Vandervalk, M.A.; Robanusmaandag, E.C.; Teriele, H.P.J. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502.
- Lankas, G.R.; Wise, L.D.; Cartwright, M.E.; Pippert, T.; Umbenhauer, D.R. Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod. Toxicol. 1998, 12, 457–463.
- Ling, V. Multidrug resistance and P-glycoprotein expression. Ann. N. Y. Acad. Sci. 1987, 507, 7–8.
- Lin, J.H.; Yamazaki, M. Clinical relevance of P-glycoprotein in drug therapy. Drug Metab. Rev. 2003, 35, 417–454.
- Karthika, C.; Sureshkumar, R. P-Glycoprotein Efflux Transporters and Its Resistance Its Inhibitors and Therapeutic Aspects. In Biomarkers and Bioanalysis Overview; IntechOpen: Rijeka, Croatia, 2020; pp. 1–12.
- Romermann, K.; Bankstahl, J.; Loscher, W.; Bankstahl, M. Pilocarpine-induced convulsive activity is limited by multidrug transporters at the rodent blood-brain barrier. J. Pharmacol. Exp. Ther. 2015, 353, 351–359.
- Kanado, Y.; Tsurudome, Y.; Omata, Y.; Yasukochi, S.; Kusunose, N.; Akamine, T.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Estradiol regulation of P-glycoprotein expression in mouse kidney and human tubular epithelial cells, implication for renal clearance of drugs. Biochem. Biophys. Res. Commun. 2019, 519, 613–619.
- Agarwal, S.; Sane, R.; Gallardo, J.L.; Ohlfest, J.R.; Elmquist, W.F. Distribution of gefitinib to the bran is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J. Pharmacol. Exp. Ther. 2010, 334, 147–155.
- Kusuhara, H.; Suzuki, H.; Sugiyama, Y. The role of P-glycoprotein in the liver. Nihon. Rinsho 1997, 55, 1069–1076.
- Lee, C.H.; Bradley, G.; Zhang, J.T.; Ling, V. Differential expression of P-glycoprotein genes in primary rat hepatocyte culture. J. Cell. Physiol. 1993, 157, 392–402.
- Kong, L.; Shen, G.; Wang, Z.; Zhuang, X.-M.; Xiao, W.-B.; Yuan, M.; Gong, Z.-H.; Li, H. Inhibition of P-Glycoprotein and multidrug resistance-associated protein 2 regulates the hepatobiliary excretion and plasma exposure of thienorphine and its glucuronide conjugate. Front. Pharmacol. 2016, 7, 242.
- Zhang, Y.; Wang, C.; Liu, Z.; Meng, Q.; Huo, X.; Liu, Q.; Sun, P.; Yang, X.; Sun, H.; Ma, X.; et al. P-gp is involved in the intestinal absorption and biliary excretion of afatinib in vitro and in rats. Pharmacol. Rep. 2018, 70, 243–250.
- Sparreboom, A.; van Asperen, J.; Mayer, U.; Schinkel, A.H.; Smit, J.W.; Meijer, D.K.F.; Borst, P.; Nooijen, W.J.; Beijnen, J.H.; van Tellingen, O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 1997, 94, 2031–2035.
- Lin, J.H. How significant is the role of P-glycoprotein in drug absorption and brain uptake? Drugs Today 2004, 40, 5–22.
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab. Dispos. 2014, 42, 623–631.
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Oriv, G. Advances in understanding the Role of P-gp in doxorubicin resistance: Molecular Pathways, therapeutic strategies, and prospects. Drug Discov. Today 2022, 27, 436–455.
- Lund, M.; Petersen, T.S.; Dalhoff, K.P. Clinical Implications of P-Glycoprotein Modulation in Drug-Drug Interactions. Drugs. 2017, 77, 859–883.
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461.
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389.
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96.
- Ahmed Juvale, I.I.; Abdul Hamid, A.A.; Abd Halim, K.B.; Che Has, A.T. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777.
- Li, Y.; Yuan, H.; Yang, K.; Xu, W.; Tang, W.; Li, X. The structure and functions of P-glycoprotein. Curr. Med. Chem. 2010, 17, 786–800.
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science. 2018, 359, 915–919.
- Sauna, Z.; Kim, I.; Nandigama, K.; Kopp, S.; Chiba, P.; Ambudkar, S.V. Catalytic cycle of ATP hydrolysis by P-glycoprotein: Evidence for formation of the E.S reaction intermediate with ATP-gamma-S, a nonhydrolyzable analogue of ATP. Biochemistry. 2007, 46, 13787–13799.
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46.
- Lagares, L.M.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure-Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362.
- Prajapati, R.; Singh, U.; Patil, A.; Khomane, K.S.; Bagul, P.; Bansal, A.K.; Sangamwar, A.T. In silico model for P-glycoprotein substrate prediction: Insights from molecular dynamics and in vitro studies. J. Comput. Aided Mol. Des. 2013, 27, 347–363.
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058.
- Sharom, F.J. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front. Oncol. 2014, 4, 41.
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure–activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228.
- Klepsch, F.; Vasanthanathan, P.; Ecker, G.F. Ligand and structure-based classification models for prediction of P-glycoprotein inhibitors. J. Chem. Inf. Model. 2014, 54, 218–229.
- Wise, J.G.; Nanayakkara, A.K.; Aljowni, M.; Chen, G.; Oliveira, M.C.D.; Ammerman, L.; Olengue, K.; Lippert, A.; Vogel, P. Optimizing Targeted Inhibitors of P-Glycoprotein Using Computational and Structure-Guided Approaches. J. Med. Chem. 2019, 62, 10645–10663.
- Fang, Y.; Liang, F.; Xia, M.; Cao, W.; Pan, S.; Wu, T.; Xu, X. Structure-activity relationship and mechanism of flavonoids on the inhibitory activity of P-glycoprotein (P-gp)-mediated transport of rhodamine123 and daunorubicin in P-gp overexpressed human mouth epidermal carcinoma (KB/MDR) cells. Food Chem. Toxicol. 2021, 155, 112381.
- Silva, V.; Gil-Martins, E.; Silva, B.; Rocha-Pereira, C.; Sousa, M.E.; Remião, F.; Silva, R. Xanthones as P-glycoprotein modulators and their impact on drug bioavailability. Expert Opin. Drug Metab. Toxicol. 2021, 17, 441–482.
- Jia, L.; Gao, X.; Fang, Y.; Zhang, H.; Wang, L.; Tang, X.; Yang, J.; Wu, C. TM2, a novel semi-synthetic taxoid, exerts anti-MDR activity in NSCLC by inhibiting P-gp function and stabilizing microtubule polymerization. Apoptosis. 2022, 27, 1015–1030.
- Hagenbuch, B.; Gao, B.; Meier, P.J. Transport of Xenobiotics Across the Blood-Brain Barrier. News Physiol. Sci. 2002, 17, 231–234.
- Chin, J.E.; Soffir, R.; Noonan, K.E.; Kyunghee, C.; Roninson, I.B. Structure and expression of the human MDR (P-glycoprotein) gene family. Mol. Cell. Biol. 1989, 9, 3808–3820.
- Ueda, K.; Clark, D.P.; Chen, C.J.; Roninson, I.B.; Gottesman, M.M.; Pastan, I. The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J. Biol. Chem. 1987, 262, 505–508.
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427.
- Rothenberg, M.L.; Mickley, L.A.; Cole, D.E.; Balis, F.M.; Tsuruo, T.; Poplack, D.G.; Fojo, A.T. Expression of the mdr-1/P-170 gene in patients with acute lymphoblastic leukemia. Blood 1989, 74, 1388–1395.
- Raguz, S.; Tamburo De Bella, M.; Tripuraneni, G.; Slade, M.J.; Higgins, C.F.; Coombes, R.C.; Yague, E. Activation of the MDR1 upstream promoter in breast carcinoma as a surrogate for metastatic invasion. Clin. Cancer Res. 2004, 10, 2776–2783.
- Cornwell, M.M.; Smith, D.E. SP1 activates the MDR1 promoter through one of two distinct G-rich regions that modulate promoter activity. J. Biol. Chem. 1993, 268, 19505–19511.
- Sauna, Z.E.; Kim, I.-W.; Ambudkar, S.V. Genomics and the mechanism of P-glycoprotein (ABCB1). J. Bioenerg. Biomembr. 2007, 39, 481–487.
- Schwab, M.; Eichelbaum, M.; Fromm, M.F. Genetic polymorphisms of the human MDR1 drug transporter. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 285–307.
- Pauli-Magnus, C.; Kroetz, D.L. Functional implications of genetic polymorphisms in the multidrug resistance gene MDR1 (ABCB1). Pharm. Res. 2004, 21, 904–913.
- Kimchi-Sarfaty, C.; Gribar, J.J.; Gottesman, M.M. Functional characterization of coding polymorphisms in the human MDR1 gene using a vaccinia virus expression system. Mol. Pharmacol. 2002, 62, 1–6.
- Speidel, J.T.; Xu, M.; Abdel-Rahman, S.Z. Promoter Haplotypes of the ABCB1 Gene Encoding the P-Glycoprotein Differentially Affect Its Promoter Activity by Altering Transcription Factor Binding. DNA Cell Biol. 2018, 37, 973–981.
- Kitada, K.; Yamasaki, T.; Aikawa, S. Amplification of the ABCB1 region accompanied by a short sequence of 200bp from chromosome 2 in lung cancer cells. Cancer Genet. Cytogenet. 2009, 194, 4–11.
- Yague, E.; Armesilla, A.L.; Harrison, G.; Elliott, J.; Sardini, A.; Higgins, C.F.; Raguz, S. P-glycoprotein (MDR1) expression in leukemic cells is regulated at two distinct steps, mRNA stabilization and translational initiation. J. Biol. Chem. 2003, 278, 10344–10352.
- Shtil, A.A. The development of multidrug resistance as an urgent cell response to exogenous effects. Biol. Membr. 2003, 20, 236–243.
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Regulation exerted by miRNAs in the promoter and UTR sequences: MDR1/P-gp expression as a particular case. DNA Cell Biol. 2012, 31, 1358–1364.
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159.
- Feng, D.D.; Zhang, H.; Zhang, P.; Zheng, Y.S.; Zhang, X.J.; Han, B.W.; Luo, X.Q.; Xu, L.; Zhou, H.; Qu, L.H.; et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J. Cell Mol. Med. 2011, 15, 2164–2175.
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503.
- Ikemura, K.; Yamamoto, M.; Miyazaki, S.; Mizutani, H.; Iwamoto, T.; Okuda, M. MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol. Pharmacol. 2013, 83, 399–405.
- Boyerinas, B.; Park, S.M.; Murmann, A.E.; Gwin, K.; Montag, A.G.; Zillhardt, M.; Hua, Y.J.; Lengyel, E.; Peter, M.E.; Boyerinas, B. Let- 7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int. J. Cancer 2012, 130, 1787–1797.
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.G.; Yang, J.M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588.
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science. 2020, 367, eaau6977.
- Zhou, H.L.; Zheng, Y.J.; Cheng, X.Z.; Lv, Y.S.; Gao, R.; Mao, H.P.; Chen, Q. Intercellular transfer of P-glycoprotein from the drug-resistant human bladder cancer cell line BIU-87 does not require cell-to-cell contact. J. Urol. 2013, 190, 1069–1075.
- Levchenko, A.; Mehta, B.M.; Niu, X.; Kang, G.; Villafania, L.; Way, D.; Polycarpe, D.; Sadelain, M.; Larson, S.M. Intercellular transfer of P-glycoprotein mediates acquired multidrug resistance in tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1933–1938.
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert. Opin. Ther. Pat. 2019, 29, 455–461.
- Yakusheva, E.N.; Shulkin, A.V.; Popova, N.M.; Chernyh, I.V.; Titov, D.S. Structure, functions, of P-glycoprotein and its role in rational pharmakoterapy. Rev. Clin. Pharmacol. Drug Ther. 2014, 12, 3–11.
- Varma, M.V.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359.
- Martin, C.; Berridge, G.; Higgins, C.F.; Mistry, P.; Charlton, P.; Callaghan, R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 2000, 58, 624–632.
- Ferreira, R.J.; Ferreira, M.J.; dos Santos, D.J. Molecular docking characterizes sub-strate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760.
- Bocci, G.; Moreau, A.; Vayer, P.; Denizot, C.; Fardel, O.; Parmentier, Y. New insights in the in vitro characterisation and molecular modelling of the P-glycoprotein inhibitory promiscuity. Europ. J. Pharmac. Sci. 2018, 121, 85–94.
- Rautio, J.; Humphreys, J.E.; Webster, L.O.; Balakrishnan, A.; Keogh, J.P.; Kunta, J.R.; Serabjit-Singh, C.J.; Polli, J.W. In vitro P-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: A recommendation for probe substrates. Drug Metab. Dispos. 2006, 34, 786–792.
- Ushigome, F.; Takanaga, H.; Matsuo, H.; Yanai, S.; Tsukimori, K.; Nakano, H.; Uchiumi, T.; Nakamura, T.; Kuwano, M.; Ohtani, H.; et al. Human placental transport of vinblastine, vincristine, digoxin and progesterone: Contribution of P-glycoprotein. Eur. J. Pharmacol. 2000, 408, 1–10.
- Prakash, A.S. Selecting surfactants for the maximum inhibition of the activity of the multidrug resistance efflux pump transporter, P-glycoprotein: Conceptual development. J. Excip. Food Chem. 2010, 1, 51–59.
- Vilas-Boas, V.; Silva, R.; Nunes, C.; Reis, S.; Ferreira, L.; Vieira, C.; Carvalho, F.; Bastos, M.L.; Remiao, F. Mechanisms of P-gp inhibition and effects on membrane fluidity of a new rifampicin derivative, 1,8-dibenzoyl-rifampicin. Toxicol. Lett. 2013, 220, 259–266.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
731
Revisions:
2 times
(View History)
Update Date:
26 Feb 2024
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No