 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudio Tirelli | -- | 3811 | 2024-02-21 07:48:14 | | | |
| 2 | Sirius Huang | Meta information modification | 3811 | 2024-02-22 02:37:24 | | |
Video Upload Options
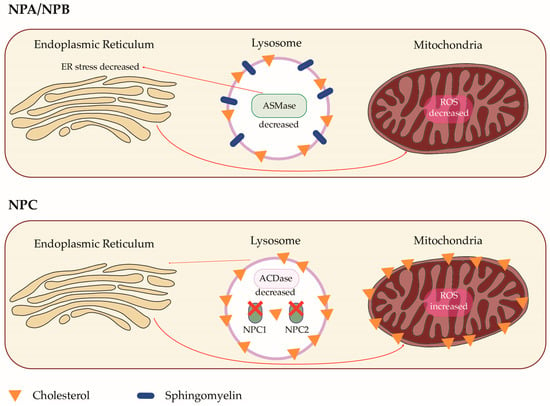
Niemann–Pick Disease (NPD) is a rare autosomal recessive disease belonging to lysosomal storage disorders. Three types of NPD have been described: NPD type A, B, and C. NPD type A and B are caused by mutations in the gene SMPD1 coding for sphingomyelin phosphodiesterase 1, with a consequent lack of acid sphingomyelinase activity. These diseases have been thus classified as acid sphingomyelinase deficiencies (ASMDs). NPD type C is a neurologic disorder due to mutations in the genes NPC1 or NPC2, causing a defect of cholesterol trafficking and esterification. Although all three types of NPD can manifest with pulmonary involvement, lung disease occurs more frequently in NPD type B, typically with interstitial lung disease, recurrent pulmonary infections, and respiratory failure.
1. Introduction
| ASMD Type A | ASMD Type B | NPD Type C | |
|---|---|---|---|
| Age of onset | Childhood | Childhood and adulthood | Mainly childhood, adulthood (underecognized) |
| Lung involvement | Present | Frequent | Rare |
| Hepato-splenomegaly | Frequent | Frequent | Frequent |
| Liver disease | Present | Frequent | Present |
| Neurodegeneration | Frequent and rapidly progressive | Present and slowly progressive | Present and rapidly progressive (in younger patients) |
| Atherosclerosis | Present | Frequent | Not described |
| Growth delay | Frequent | Frequent | Not frequently described |
| Hypotonia | Frequent | Rare | Present |
2. The Genetic Basis of Niemann–Pick Disease Type A and B: Disease-Causing Mutations and Genotype–Phenotype Relationships
3. Acid Sphingomyelinase Deficiencies: Clinical Aspects in Niemann–Pick Disease Type A and B
3.1. Niemann–Pick Disease Type A (ASMD Type A)
3.2. Niemann–Pick Disease Type B (ASMD Type B)
3.3. Niemann–Pick Disease Type A/B (ASMD Type A/B)
4. The Genetic Basis of Niemann–Pick Disease Type C: Disease-Causing Mutations and Genotype–Phenotype Relationships
5. Niemann–Pick Disease Type C: Clinical Aspects
References
- Takahashi, T.; Suchis, M.; Desnicks, R.J.; Takadag, G.; Schuchmanst, E.H. Identification and Expression of Five Mutations in the Human Acid Sphingomyelinase Gene Causing Types A and B Niemann-Pick Disease. Molecular Evidence for Genetic Heterogeneity in the Neuronopathic and Non-Neuronopathic Forms. J. Biol. Chem. Med. Mol. Genet. 1992, 267, 12552–12558.
- Wang, R.; Qin, Z.; Huang, L.; Luo, H.; Peng, H.; Zhou, X.; Zhao, Z.; Liu, M.; Yang, P.; Shi, T. SMPD1 expression profile and mutation landscape help decipher genotype–phenotype association and precision diagnosis for acid sphingomyelinase deficiency. Hereditas 2023, 160, 11.
- Zampieri, S.; Filocamo, M.; Pianta, A.; Lualdi, S.; Gort, L.; Coll, M.J.; Sinnott, R.; Geberhiwot, T.; Bembi, B.; Dardis, A. SMPD1 Mutation Update: Database and Comprehensive Analysis of Published and Novel Variants. Hum. Mutat. 2016, 37, 139–147.
- Dardis, A.; Zampieri, S.; Filocamo, M.; Burlina, A.; Bembi, B.; Pittis, M.G. Functional in vitro characterization of 14 SMPD1 mutations identified in Italian patients affected by Niemann-Pick Type B disease. Hum. Mutat. 2005, 26, 164.
- Tóth, B.; Erdős, M.; Székely, A.; Ritli, L.; Bagossi, P.; Sümegi, J.; Maródi, L. Molecular genetic characterization of novel sphingomyelin phosphodiesterase 1 mutations causing Niemann-Pick Disease. JIMD Rep. 2012, 3, 125–129.
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Type A Niemann-Pick Disease: A frameshift mutation in the acid sphingomyelinase gene (fsP330) occurs in Ashkenazi Jewish patients. Hum. Mutat. 1993, 2, 317–319.
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Identification and Expression of a Common Missense Mutation (L302P) in the Acid Sphingomyelinase Gene of Ashkenazi Jewish Type A Niemann-Pick Disease Patients. Blood 1992, 80, 2081–2087.
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Niemann-Pick type B disease. Identification of a single codon deletion in the acid sphingomyelinase gene and genotype/phenotype correlations in type A and B patients. J. Clin. Investig. 1991, 88, 806–810.
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Niemann-Pick Disease: A frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc. Natl. Acad. Sci. USA 1991, 88, 3748–3752.
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982.
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick Disease. Mol. Genet. Metab. 2017, 120, 27–33.
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The demographics and distribution of type B Niemann-Pick Disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419.
- McGovern, M.M.; Avetisyan, R.; Sanson, B.J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41.
- Rossi, G.; Cavazza, A.; Spagnolo, P.; Bellafiore, S.; Kuhn, E.; Carassai, P.; Caramanico, L.; Montanari, G.; Cappiello, G.; Nannini, N.; et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series ‘Pathology for the clinician’ Edited by Peter Dorfmüller and Alberto Cavazza. Eur. Respir. Rev. 2017, 26, 170009.
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of Type A Niemann-Pick Disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232.
- Ospina, L.H.; Lyons, C.J.; McCormick, A.Q. ‘Cherry-red spot’ or ‘perifoveal white patch’? Can. J. Ophthalmol. 2005, 40, 609–610.
- Leavitt, J.A.; Kotagal, S. The ‘cherry red’ spot. Pediatr. Neurol. 2007, 37, 74–75.
- McGovern, M.M.; Wasserstein, M.P.; Giugliani, R.; Bembi, B.; Vanier, M.T.; Mengel, E.; Brodie, S.E.; Mendelson, D.; Skloot, G.; Cox, G.F.; et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick Disease type B. Pediatrics 2008, 122, e341–e349.
- Simpson, W.L.; Mendelson, D.; Wasserstein, M.P.; McGovern, M.M. Imaging manifestations of Niemann-Pick Disease type B. AJR. Am. J. Roentgenol. 2010, 194, W12–W19.
- Wasserstein, M.P.; Larkin, A.E.; Glass, R.B.; Schuchman, E.H.; Desnick, R.J.; McGovern, M.M. Growth restriction in children with type B Niemann-Pick Disease. J. Pediatr. 2003, 142, 424–428.
- Wasserstein, M.P.; Aron, A.; Brodie, S.E.; Simonaro, C.; Desnick, R.J.; McGovern, M.M. Acid sphingomyelinase deficiency: Prevalence and characterization of an intermediate phenotype of Niemann-Pick Disease. J. Pediatr. 2006, 149, 554–559.
- McGovern, M.M.; Wasserstein, M.P.; Aron, A.; Desnick, R.J.; Schuchman, E.H.; Brodie, S.E. Ocular manifestations of Niemann-Pick Disease type B. Ophthalmology 2004, 111, 1424–1427.
- Gülhan, B.; Özçelik, U.; Gürakan, F.; Güçer, Ş.; Orhan, D.; Cinel, G.; Yalcin, E.; Ersoz, D.D.; Kiper, N.; Kale, G.; et al. Different features of lung involvement in Niemann-Pick Disease and Gaucher disease. Respir. Med. 2012, 106, 1278–1285.
- Vance, J.E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006, 580, 5518–5524.
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Duthel, S.; Harzer, K.; Pentchev, P.G.; Revol, A.; Louisot, P. Type C Niemann-Pick Disease: Biochemical aspects and phenotypic heterogeneity. Dev. Neurosci. 1991, 13, 307–314.
- Greer, W.L.; Riddell, D.C.; Gillan, T.L.; Girouard, G.S.; Sparrow, S.M.; Byers, D.M.; Dobson, M.J.; Neumann, P.E. The Nova Scotia (type D) form of Niemann-Pick Disease is caused by a G3097-->T transversion in NPC1. Am. J. Hum. Genet. 1998, 63, 52–54.
- Verot, L.; Chikh, K.; Freydière, E.; Honoré, R.; Vanier, M.T.; Millat, G. Niemann-Pick C disease: Functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin. Genet. 2007, 71, 320–330.
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 disease: The I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329.
- Ramirez, C.M.; Lopez, A.M.; Le, L.Q.; Posey, K.S.; Weinberg, A.G.; Turley, S.D. Ontogenic changes in lung cholesterol metabolism, lipid content, and histology in mice with Niemann-Pick type C disease. Biochim. Biophys. Acta 2014, 1841, 54–61.
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001, 68, 1373–1385.
- Greer, W.L.; Dobson, M.J.; Girouard, G.S.; Byers, D.M.; Riddell, D.C.; Neumann, P.E. Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am. J. Hum. Genet. 1999, 65, 1252–1260.
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Snow, K.; et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am. J. Hum. Genet. 2001, 68, 1361–1372.
- Park, W.D.; O’Brien, J.F.; Lundquist, P.A.; Kraft, D.L.; Vockley, C.W.; Karnes, P.S.; Patterson, M.C.; Snow, K. Identification of 58 novel mutations in Niemann-Pick Disease type C: Correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum. Mutat. 2003, 22, 313–325.
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301.
- Millat, G.; Chikh, K.; Naureckiene, S.; Sleat, D.E.; Fensom, A.H.; Higaki, K.; Elleder, M.; Lobel, P.; Vanier, M.T. Niemann-Pick Disease type C: Spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am. J. Hum. Genet. 2001, 69, 1013–1021.
- Vanier, M.T. Niemann-Pick Disease type C. Orphanet J. Rare Dis. 2010, 5, 1.
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; International Niemann-Pick Disease Registry (INPDR); et al. Consensus clinical management guidelines for Niemann-Pick Disease type C. Orphanet J. Rare Dis. 2018, 13, 50.
- Vo, M.L.; Levy, T.; Lakhani, S.; Wang, C.; Ross, M.E. Adult-onset Niemann-Pick Disease type C masquerading as spinocerebellar ataxia. Mol. Genet. Genom. Med. 2022, 10, 1906.
- Patterson, M.C. A riddle wrapped in a mystery: Understanding Niemann-Pick Disease, type C. Neurologist 2003, 9, 301–310.
- Vanier, M.T.; Millat, G. Niemann-Pick Disease type C. Clin. Genet. 2003, 64, 269–281.
- Wraith, J.E.; Baumgartner, M.R.; Bembi, B.; Covanis, A.; Levade, T.; Mengel, E.; Pineda, M.; Sedel, F.; Topcu, M.; Patterson, M.C.; et al. Recommendations on the diagnosis and management of Niemann-Pick Disease type C. Mol. Genet. Metab. 2009, 98, 152–165.
- Guillemot, N.; Troadec, C.; De Villemeur, T.B.; Clément, A.; Fauroux, B. Lung disease in Niemann-Pick Disease. Pediatr. Pulmonol. 2007, 42, 1207–1214.
- Griese, M.; Brasch, F.; Aldana, V.R.; Cabrera, M.M.; Goelnitz, U.; Ikonen, E.; Karam, B.J.; Liebisch, G.; Linder, M.D.; Lezana, F.J.; et al. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin. Genet. 2010, 77, 119–130.

