Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Salcedo Betancourt | -- | 1695 | 2024-02-20 18:28:46 | | | |
| 2 | Jessie Wu | Meta information modification | 1695 | 2024-02-21 02:05:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Salcedo-Betancourt, J.D.; Moe, O.W. Acid–Base Homeostasis and Renal Calcium–Phosphate Handling. Encyclopedia. Available online: https://encyclopedia.pub/entry/55247 (accessed on 08 August 2026).
Salcedo-Betancourt JD, Moe OW. Acid–Base Homeostasis and Renal Calcium–Phosphate Handling. Encyclopedia. Available at: https://encyclopedia.pub/entry/55247. Accessed August 08, 2026.
Salcedo-Betancourt, Juan D., Orson W. Moe. "Acid–Base Homeostasis and Renal Calcium–Phosphate Handling" Encyclopedia, https://encyclopedia.pub/entry/55247 (accessed August 08, 2026).
Salcedo-Betancourt, J.D., & Moe, O.W. (2024, February 20). Acid–Base Homeostasis and Renal Calcium–Phosphate Handling. In Encyclopedia. https://encyclopedia.pub/entry/55247
Salcedo-Betancourt, Juan D. and Orson W. Moe. "Acid–Base Homeostasis and Renal Calcium–Phosphate Handling." Encyclopedia. Web. 20 February, 2024.
Copy Citation
Both calcium and phosphate metabolism are involved in acid–base homeostasis at several physiological intersections. Phosphate plays a key role in defense against metabolic acidosis, both as an intracellular and extracellular buffer, as well as in the renal excretion of excess H+ in the form of urinary titratable acid through this buffering reaction (Na2HPO4 ⇌ HPO42−+ 2Na and HPO42− + H+ ⇌ H2PO4−). The skeleton acts as an extracellular buffer in states of metabolic acidosis, as the acid-induced dissolution of bone hydroxyapatite releases Ca2+ and phosphate into the extracellular fluid (ECF).
phosphate
calcium
acidosis
1. Normal Renal Calcium and Phosphate Handling
Total body calcium stores are approximately 1000 g (25,000 mmol), mostly in the form of hydroxyapatite in the mineralized bone matrix (99%) [1]. Calcium homeostasis is balanced through three organ systems: intestinal, bone and kidney. In the kidney, 60–70% of filtered calcium is passively reabsorbed with sodium and water in the proximal tubule, and 20% in the thick ascending limb, both via paracellular mechanisms, and the remaining 10–20% is reabsorbed transcellularly through the apical calcium channel TRPV5 and extruded through the basolateral Na-Ca exchanger and Ca-ATPase, in the distal tubule and collecting duct (Figure 1). The regulatory mechanisms of renal calcium reabsorption adapt to the body’s needs, with reabsorption increased by hypocalcemia, PTH release and volume contraction [2]. Plasma ionized calcium is the main regulator of PTH secretion, where in the setting of hypercalcemia, CaSR is stimulated, which inhibits PTH secretion on parathyroid chief cells. PTH secretion enhances bone turnover through osteoblast activation and stimulates the 1α-hydroxylase conversion of 25(OH)D3 into 1,25(OH)2D3, which, in turn, increases the intestinal absorption of calcium. Additionally, PTH production is inhibited by 1,25(OH)2D3 [3]. At the tubular level, PTH enhances distal calcium reabsorption through the upregulation of TRPV5, leading to increased serum calcium levels [4].
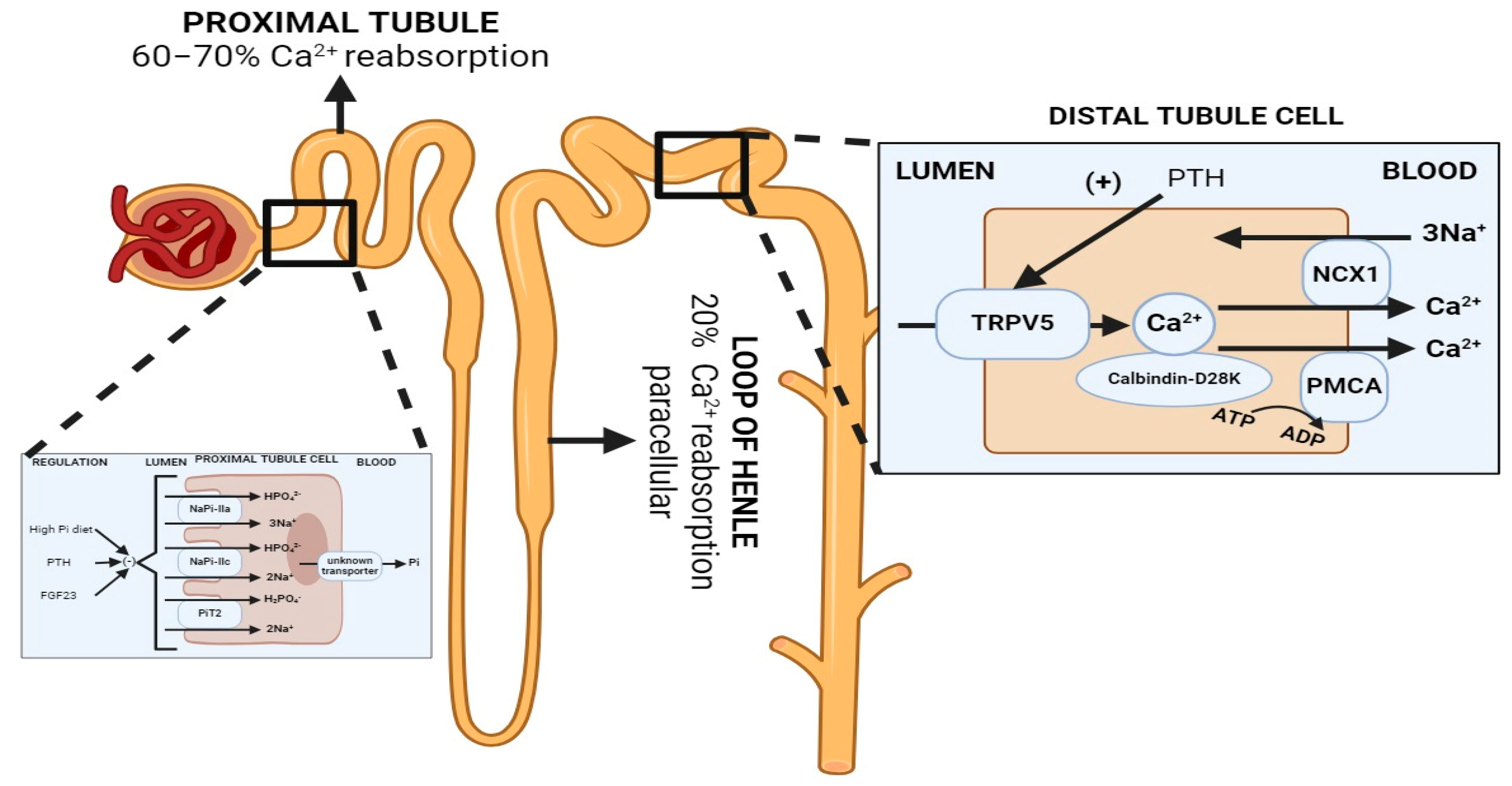
Figure 1. Normal renal handling of phosphate and calcium. Left: Phosphate handling. Phosphate is predominantly reabsorbed in the proximal tubule. PTH and FGF-23 promote the retrieval and degradation of NaPi-IIa (Slc34A1), NaPi-IIc (Slc34A3) and PiT2 (Slc20A2). Calcitriol is believed to increase phosphate reabsorption in the proximal tubule, but the effects are confounded since it also increases PTH levels. Right: Calcium handling. 60–70% of filtered calcium is passively reabsorbed with sodium and water in the proximal tubule, and 20% in the thick ascending limb via paracellular mechanisms, and 10–20% is reabsorbed transcellularly through TRPV5, an apical membrane calcium channel in the distal tubule and collecting duct. PTH enhances distal calcium reabsorption through the upregulation of TRPV5, leading to increased serum calcium levels. Abbreviations: Pi = inorganic phosphate, NCX = Na+/Ca2+ exchanger, PMCA = plasma membrane calcium ATPase. Created with BioRender.com, accessed on 23 December 2023.
Phosphate is essential for many physiological needs, including cellular energy production, cell signaling and structure, oxygen delivery to tissues and bone mineralization, among others. Most phosphate exists as hydroxyapatite in the mineralized bone matrix (85%), and 14% intracellularly [2], of which 70% occurs as organic (phospholipids, phosphoproteins, nucleic acids, adenosine triphosphate (ATP) and cyclic adenosine monophosphate (cAMP)) and 30% as inorganic phosphate. Only 1% of total body phosphate exists as non-osseous extracellular serum phosphate [4] in the serum, of which 15% is found to be protein bound, 14% is complexed to cations [3], and 47% is ionized as divalent (HPO42−) and monovalent (H2PO4−) phosphate in a 4:1 ratio at pH 7.4 [4].
Phosphate homeostasis is balanced through three major systems: intestinal uptake, bone release/incorporation and renal excretion. Systemic phosphate balance is mainly regulated through changes in the urinary fractional excretion of phosphate under typical conditions [5][6]. Urinary phosphate contributes to 50% of titratable acids. Hence, renal phosphate excretion plays a significant role in renal net acid excretion (NAE) [7]. Only the ingestion of divalent phosphate (HPO42−) and the urinary excretion of monovalent phosphate (H2PO4−) represents a net loss of H+ and a net gain of HCO3−; in contrast, the ingestion and excretion of monovalent phosphate (H2PO4−), does not represent either a gain or loss of base [8].
Phosphate is freely filtered by the glomerulus (about 90% of plasma phosphorus). Then, 80–90% of renal phosphate is reabsorbed in the proximal tubule. Reabsorption is sodium-dependent as it is performed by the sodium–phosphate co-transporters NaPi-IIa (Slc34A1) and NaPi-IIc (Slc34A3) located in the proximal tubule apical membrane [9]. Hence, renal phosphate excretion is determined by the glomerular filtration rate and the tubular maximum reabsorption rate (TMP-phosphate). Phosphaturia occurs when maximal tubular phosphate reabsorption becomes saturated. The regulatory mechanisms of renal phosphate reabsorption adapt to the body’s needs, [4] including the parathyroid hormone (PTH), Fibroblast Growth Factor-23 (FGF-23) and αKlotho [10]. FGF-23 and PTH promote the retrieval and degradation of NaPi-IIa [11], NaPi-Iic [9][12] and PiT2 (Figure 1) [13]. Additionally, in a phosphate depletion state, there is a reduction in the phosphaturic effect of PTH [4] due to the augmentation of the tubular reabsorption of phosphate [14]. It has been recently described that the calcium-sensing receptor (CaSR) represents a phosphate-sensing mechanism in the parathyroid gland, since raising the extracellular phosphate concentration significantly inhibited CaSR activity via non-competitive antagonism and stimulated PTH secretion in murine models [15].
2. Renal Calcium Handling in Metabolic Acidosis
Metabolic acidosis increases urinary calcium excretion, leading to negative calcium balance when persistent [16]. There are several proposed mechanisms to explain the hypercalciuric effect of metabolic acidosis (Figure 2) [17]: (1) the decreased electrochemical gradient required for paracellular calcium reabsorption in the proximal tubule, (2) the decreased driving force and permeability for paracellular calcium reabsorption in the thick ascending limb (TAL) via the claudin 16/19 complex [18], and (3) the direct inhibitory effect on TRPV5 gating caused by the acidification of either extracellular or intracellular pH.
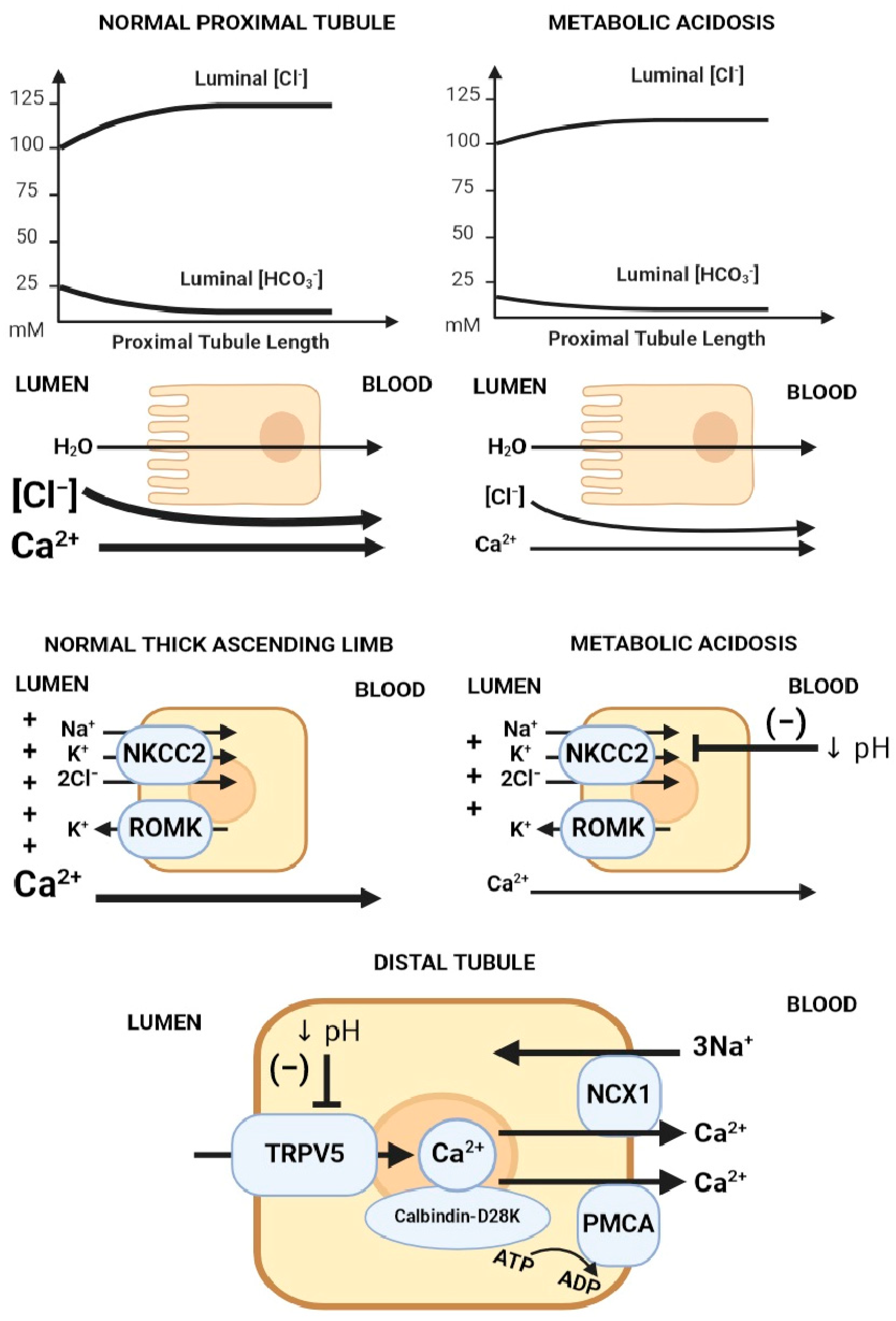
Figure 2. Mechanisms of acidosis-induced hypercalciuria. Proposed mechanisms of hypercalciuria in metabolic acidosis axially down the nephron: (1) Decreased electrochemical gradient required for paracellular calcium reabsorption in the proximal tubule. Luminal calcium is concentrated through the isotonic absorption of NaCl and water. Removal of HCO3− from the lumen increases luminal [Cl−], creating an electrochemical gradient for paracellular calcium reabsorption. In metabolic acidosis, there is lower luminal HCO3−, leading to a blunted rise in luminal [Cl−], which translates into a lower chemical driving force for calcium reabsorption. (2) The suppression of Na/K/2Cl transport decreases the electrical gradient required for paracellular calcium reabsorption in the thick ascending limb (TAL). (3) TRPV5 gating inhibition caused by acidification of either extracellular or intracellular pH. Abbreviations: ↓ pH = Decreased pH. Created with BioRender.com, accessed on 23 December 2023.
Calcium reabsorption by the proximal tubule is carried primarily via the paracellular route, driven electrochemically by the luminal calcium concentration due to isotonic water and solute (mainly sodium chloride) absorption; hence, the main regulator of calcium flux in the proximal tubule is sodium chloride/water reabsorption. In metabolic acidosis, there is a blunted rise in luminal chloride compared to when serum bicarbonate is normal, which translates into a lower driving force for calcium absorption. In the thick ascending limb, calcium is absorbed primarily via the paracellular route, driven by positive luminal voltage (generated via sodium reabsorption via NKCC2 (Na-K-2Cl cotransporter) and potassium recycling via ROMK). Metabolic acidosis inhibits apical NaCl absorption via NKCC2, leading to reduced luminal voltage and decreased paracellular calcium reabsorption at the TAL segment. On the other hand, calcium reabsorption in the distal tubule mainly occurs via the transcellular route through the TRPV5 calcium apical calcium channel, after which calcium binds calbindin-D28K in the cytosol, which facilitates diffusion through the cytosol and eventual extrusion across sodium/calcium exchangers (NCX1) and calcium/ATPase (PMCA) on the basolateral membrane (Figure 2).
3. Renal Phosphate Handling in Metabolic Acidosis
Urinary phosphate excretion is predominantly determined by PTH and FGF-23. However, disturbances in acid–base balance also influence renal phosphate handling. During metabolic acidosis, there is a significant increase in the renal excretion of phosphate [19][20], which contributes to acid removal through titratable acid. As kidney function declines in chronic kidney disease (CKD), there is a decrease in ammonium formation and secretion by the proximal tubule [21]. There is an increase in the excretion of monovalent phosphate (H2PO4−) as a compensation through metabolic acidosis-induced phosphaturia [20]. Lower serum bicarbonate is associated with higher renal phosphate excretion in patients with CKD [22].
There are several mechanisms proposed for this effect (Figure 3): (1) titration of the substrate with H+, decreasing the luminal availability of divalent phosphate (HPO42−) for uptake; (2) the direct inhibitory gating of H+ on NaPi-IIa and NaPi-IIc [9]; (3) NaPi-IIa trafficking modifications, including the impaired delivery of NaPi-IIa to the apical BBM and/or enhanced internalization from the BBM [23]; and (4) the reduced transcription and translation of NaPi-IIa [20].
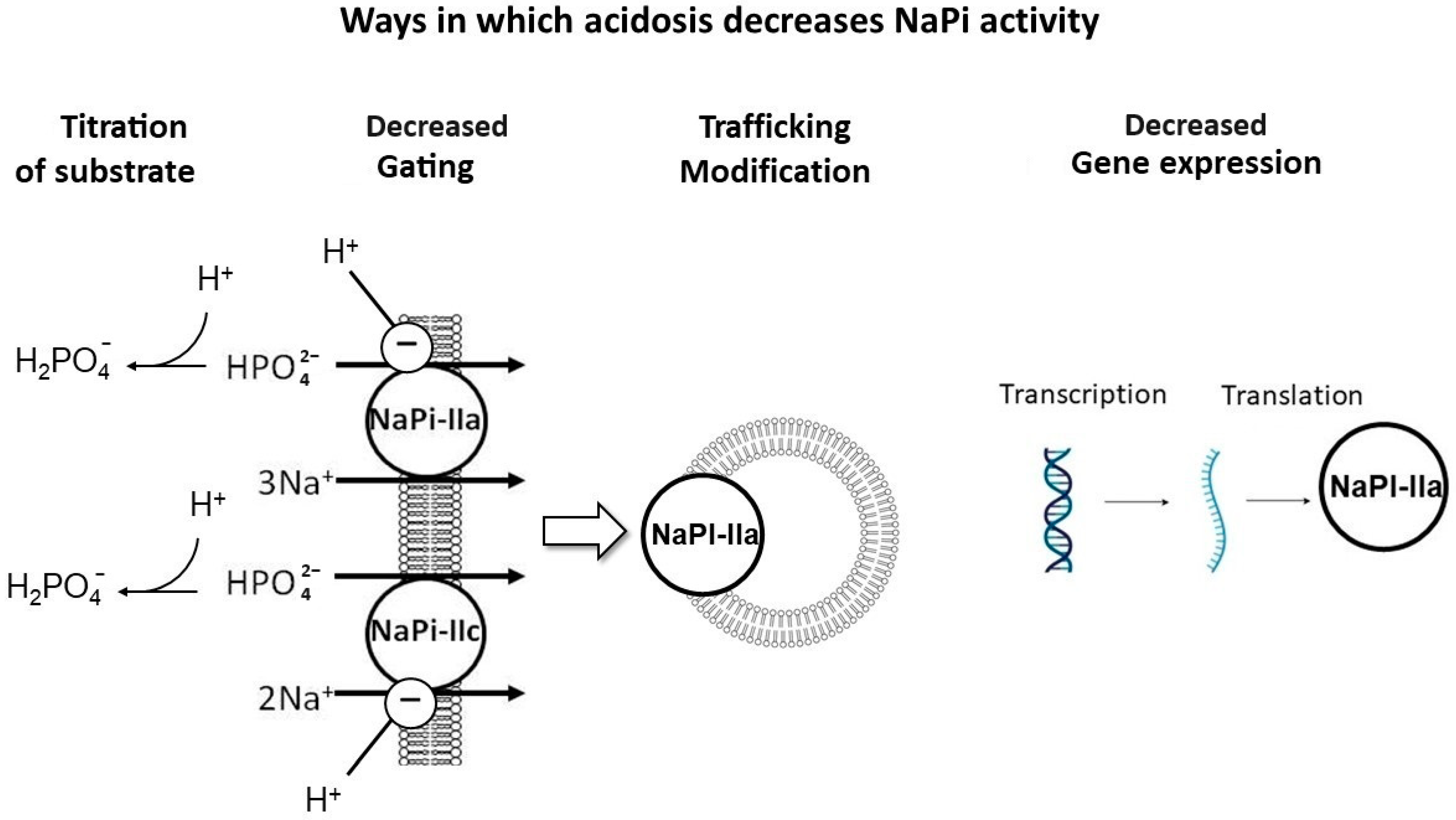
Figure 3. Mechanisms of acidosis-induced phosphaturia. Proposed mechanisms to explain the phosphaturic effect of metabolic acidosis: (1) Titration of substrate with H+, decreasing the luminal availability of divalent phosphate (HPO42−), the preferred substrate to be absorbed. (2) Direct inhibitory gating of H+ on NaPi-IIa and IIc. (3) NaPi-IIa trafficking modifications, including impaired delivery of NaPi-IIa to the apical brush border membrane (BBM) and/or enhanced internalization from the BBM. (4) Impaired transcription and translation of NaPi-IIa. Created with BioRender.com, accessed on 23 December 2023.
Metabolic acidosis has been shown to induce the expression of the phosphaturic hormones FGF-23 [24] and PTH [25][26][27][28]. Chronic metabolic acidosis for over 12 h led to the impaired transcription and translation of NaPi-IIa and decreased BBM Na–Pi cotransport activity. However, after the induction of acute metabolic acidosis (less than 6 h of acid loading), there was decreased Na-Pi cotransport activity, independent of changes in Slc34A1 mRNA expression [20]. The decrease in the apical NaPi-IIa protein in acute metabolic acidosis may be mediated by either the impaired delivery of NaPi-IIa to the BBM or enhanced internalization from the apical BBM [20][23]. Others have attributed acid-induced phosphaturia to the direct effects of local pH on the flux of these co-transporters in the proximal tubule [9], which was further confirmed with transcriptome analysis of acid-loaded murine kidneys that revealed a decrease in both NaPi-IIa and NaPi-IIc mRNA abundance, while the abundance of Pit1 and Pit2 transporters remained unchanged despite the induction of metabolic acidosis [9]. Nowik et al. showed that NaPi-IIa KO mice (Slc34A1−/−) had no acid-induced phosphaturia, while WT mice had a progressive reduction in BBM Na/Pi cotransport activity during ammonium chloride-induced metabolic acidosis, suggesting that metabolic acidosis-induced phosphaturia is caused by an inhibitory effect of low pH on the flux of NaPi-IIa [9].
References
- Moorthi, R.N.; Moe, S.M. CKD-mineral and bone disorder: Core curriculum 2011. Am. J. Kidney Dis. 2011, 58, 1022–1036.
- Moe, S.M. Disorders of calcium, phosphorus, and magnesium. Am. J. Kidney Dis. 2005, 45, 213–218.
- Baker, S.B.; Worthley, L.I.G. The Essentials of Calcium, Magnesium and Phosphate Metabolism: Part I. Physiology. Crit. Care Resusc. 2002, 4, 301–306.
- O’Dwyer, L. Disorders of Phosphorus. In Acid-Base and Electrolyte Handbook for Veterinary Technicians; Wiley: Hoboken, NJ, USA, 2016; pp. 66–78.
- Alizadeh Naderi, A.S.; Reilly, R.F. Hereditary disorders of renal phosphate wasting. Nat. Rev. Nephrol. 2010, 6, 657–665.
- Berndt, T.J.; Knox, F.G. Renal Regulation of Phosphate Excretion; Seldin, D.G., Giebisch, G., Eds.; Raven Press: New York, NY, USA, 1992; pp. 2511–2532.
- Wrong, O.; Davies, H.E. The excretion of acid in renal disease. Q. J. Med. 1959, 28, 259–313.
- Halperin, M.L.; Kamel, K.S.; Goldstein, M.B. Fluid, Electrolyte and Acid-Base Physiology; Elsevier: Amsterdam, The Netherlands, 2016.
- Nowik, M.; Picard, N.; Stange, G.; Capuano, P.; Tenenhouse, H.S.; Biber, J.; Murer, H.; Wagner, C.A. Renal phosphaturia during metabolic acidosis revisited: Molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch. 2008, 457, 539–549.
- Karim-Jimenez, Z.; Hernando, N.; Biber, J.; Murer, H. A dibasic motif involved in parathyroid hormone-induced down-regulation of the type IIa NaPi cotransporter. Proc. Natl. Acad. Sci. USA 2000, 97, 12896–12901.
- Miyamoto, K.; Ito, M.; Kuwahata, M.; Kato, S.; Segawa, H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther. Apher. Dial. 2005, 9, 331–335.
- Ohkido, I.; Segawa, H.; Yanagida, R.; Nakamura, M.; Miyamoto, K. Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflugers Arch. 2003, 446, 106–115.
- Blaine, J.; Chonchol, M.; Levi, M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 1257–1272.
- Chan, J.C.; Alon, U. Tubular disorders of acid-base and phosphate metabolism. Nephron 1985, 40, 257–279.
- Centeno, P.P.; Herberger, A.; Mun, H.C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 4693.
- Gafter, U.; Kraut, J.A.; Lee, D.B.; Silis, V.; Walling, M.W.; Kurokawa, K.; Haussler, M.R.; Coburn, J.W. Effect of metabolic acidosis in intestinal absorption of calcium and phosphorus. Am. J. Physiol. 1980, 239, G480–G484.
- Moe, O.W.; Huang, C.L. Hypercalciuria from acid load: Renal mechanisms. J. Nephrol. 2006, 19 (Suppl. S9), S53–S61.
- Kim, G.H. Renal Mechanisms for Hypercalciuria Induced by Metabolic Acidosis. Am. J. Nephrol. 2022, 53, 839–846.
- Schiess, W.A.; Ayer, J.L.; Lotspeich, W.D.; Pitts, R.F.; Miner, P. The Renal Regulation of Acid-Base Balance in Man. Ii. Factors Affecting the Excretion of Titratable Acid by the Normal Human Subject. J. Clin. Investig. 1948, 27, 57–64.
- Ambuhl, P.M.; Zajicek, H.K.; Wang, H.; Puttaparthi, K.; Levi, M. Regulation of renal phosphate transport by acute and chronic metabolic acidosis in the rat. Kidney Int. 1998, 53, 1288–1298.
- Kim, H.J. Metabolic Acidosis in Chronic Kidney Disease: Pathogenesis, Clinical Consequences, and Treatment. Electrolyte Blood Press 2021, 19, 29–37.
- Khairallah, P.; Isakova, T.; Asplin, J.; Hamm, L.; Dobre, M.; Rahman, M.; Sharma, K.; Leonard, M.; Miller, E., 3rd; Jaar, B.; et al. Acid Load and Phosphorus Homeostasis in CKD. Am. J. Kidney Dis. 2017, 70, 541–550.
- Villa-Bellosta, R.; Sorribas, V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch. 2010, 459, 499–508.
- Krieger, N.S.; Culbertson, C.D.; Kyker-Snowman, K.; Bushinsky, D.A. Metabolic acidosis increases fibroblast growth factor 23 in neonatal mouse bone. Am. J. Physiol. Renal Physiol. 2012, 303, F431–F436.
- Campion, K.L.; McCormick, W.D.; Warwicker, J.; Khayat, M.E.; Atkinson-Dell, R.; Steward, M.C.; Delbridge, L.W.; Mun, H.C.; Conigrave, A.D.; Ward, D.T. Pathophysiologic changes in extracellular pH modulate parathyroid calcium-sensing receptor activity and secretion via a histidine-independent mechanism. J. Am. Soc. Nephrol. 2015, 26, 2163–2171.
- Disthabanchong, S.; Martin, K.J.; McConkey, C.L.; Gonzalez, E.A. Metabolic acidosis up-regulates PTH/PTHrP receptors in UMR 106-01 osteoblast-like cells. Kidney Int. 2002, 62, 1171–1177.
- Bushinsky, D.A.; Nilsson, E.L. Additive effects of acidosis and parathyroid hormone on mouse osteoblastic and osteoclastic function. Am. J. Physiol. 1995, 269, C1364–C1370.
- Lopez, I.; Aguilera-Tejero, E.; Felsenfeld, A.J.; Estepa, J.C.; Rodriguez, M. Direct effect of acute metabolic and respiratory acidosis on parathyroid hormone secretion in the dog. J. Bone Miner. Res. 2002, 17, 1691–1700.
- Hamm, L.L.; Simon, E.E. Roles and mechanisms of urinary buffer excretion. Am. J. Physiol. 1987, 253, F595–F605.
- Emmett, M.; Seldin, D.W. Disturbances in acid-base balance during hypophosphatemia and phosphate depletion. Adv. Exp. Med. Biol. 1978, 103, 313–325.
More
Information
Subjects:
Urology & Nephrology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
21 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No