Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ROY A POBLETE | -- | 3183 | 2024-02-19 23:01:50 | | | |
| 2 | Lindsay Dong | Meta information modification | 3183 | 2024-02-20 02:35:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Poblete, R.A.; Zhong, C.; Patel, A.; Kuo, G.; Sun, P.Y.; Xiao, J.; Fan, Z.; Sanossian, N.; Towfighi, A.; Lyden, P.D. Post-Traumatic Cerebral Infarction. Encyclopedia. Available online: https://encyclopedia.pub/entry/55196 (accessed on 27 June 2026).
Poblete RA, Zhong C, Patel A, Kuo G, Sun PY, Xiao J, et al. Post-Traumatic Cerebral Infarction. Encyclopedia. Available at: https://encyclopedia.pub/entry/55196. Accessed June 27, 2026.
Poblete, Roy A., Charlotte Zhong, Anish Patel, Grace Kuo, Philip Y. Sun, Jiayu Xiao, Zhaoyang Fan, Nerses Sanossian, Amytis Towfighi, Patrick D. Lyden. "Post-Traumatic Cerebral Infarction" Encyclopedia, https://encyclopedia.pub/entry/55196 (accessed June 27, 2026).
Poblete, R.A., Zhong, C., Patel, A., Kuo, G., Sun, P.Y., Xiao, J., Fan, Z., Sanossian, N., Towfighi, A., & Lyden, P.D. (2024, February 19). Post-Traumatic Cerebral Infarction. In Encyclopedia. https://encyclopedia.pub/entry/55196
Poblete, Roy A., et al. "Post-Traumatic Cerebral Infarction." Encyclopedia. Web. 19 February, 2024.
Copy Citation
Traumatic brain injury (TBI) is a common diagnosis requiring acute hospitalization. Long-term, TBI is a significant source of health and socioeconomic impact in the United States and globally. The goal of clinicians who manage TBI is to prevent secondary brain injury. In this population, post-traumatic cerebral infarction (PTCI) acutely after TBI is an important but under-recognized complication that is associated with negative functional outcomes.
traumatic brain injury
stroke
cervical artery dissection
cerebral venous thrombosis
vasospasm
1. Introduction
1.1. The Health and Socioeconomic Impact of Traumatic Brain Injury
Traumatic brain injury (TBI) is a significant cause of death and long-term disability in the United States (US) and worldwide. According to the US Centers for Disease Control and Prevention, TBI results in 2.9 million emergency room visits, 288,000 annual hospitalizations, and 57,000 TBI-related deaths [1]. In addition to high mortality rates, TBI survivors often suffer from long-term functional disability with cognitive, emotional, behavioral, and physical impairments [2]. The impact both on the individual and society at large is significant. The cumulative lifetime medical costs of TBI are estimated to be USD 76.5 billion nationwide [3][4].
1.2. Secondary Brain Injury in Traumatic Brain Injury
Primary TBI prevention has the greatest impact on the overall burden of disease; however, limiting secondary injury after TBI is a clinical priority. After the initial impact or acceleration–deceleration injury, multiple pathophysiologic mechanisms lead to secondary brain injury, including delayed hemorrhagic expansion, ischemic injury and microthrombi formation, neuroinflammation, oxidative stress, metabolic dysfunction, and cerebral edema. Neuroinflammation, which begins early after TBI, plays a central role in mediating secondary neuronal death [5]. This process involves a cascade of reactions leading to excitotoxicity from the activation of glutamate and calcium receptors, phospholipid membrane breakdown, and microglial activation mediated by chemokines and cytokines that remain elevated several months after TBI [6][7][8][9]. Inflammation also promotes blood–brain barrier (BBB) permeability, resulting in a low-resistance pathway for pro-inflammatory cytokines, macrophages, neutrophils, and water into brain parenchyma [10]. The resulting neuronal injury and cerebral edema can lead to irreversible brain injury and life-threatening herniation syndromes. These deleterious effects can be minimized with appropriate post-TBI monitoring and care.
1.3. Post-Traumatic Cerebral Infarction: An Important Cause of Secondary Brain Injury
Given the morbidity and mortality associated with TBI, it is important to recognize common causes of secondary brain injury. Although, individually, acute ischemic stroke (AIS) and TBI are extensively studied and described in the literature, less is known about the development and treatment of post-traumatic cerebral infarction (PTCI).
Delayed cerebral ischemia is a feared complication of TBI that worsens secondary brain injury and is potentially preventable. PTCI is not fully characterized; however, observational studies demonstrate its impact on patient outcomes. The true incidence of PTCI is unknown but is reported to be 2.1–12% based on retrospective reviews [11][12][13], often occurring within the first two weeks of TBI [13]. Differences in the reported incidence of PTCI are likely due to heterogenous patient populations and diagnostic criteria. Research has consistently demonstrated an association between PTCI and increased mortality [11][12][13]. In a large US secondary database analysis by Kowalski et al., AIS after TBI was found to result in poor outcomes by multiple functional measures, including a 13.3-point reduction in Functional Independence Measure total score, a 1.9-point increase in Disability Rating Scale score, and an 18-day increase in post-TBI amnesia duration [11].
The largest identified risk factors for PTCI are a low Glasgow Coma Scale (GCS) score on presentation, brain herniation, a need for decompressive craniectomy (DC), and cervical artery (carotid or vertebral artery) dissection (CAD) [11][12][13]. Although age is a significant risk factor for spontaneous AIS, its influence on PTCI incidence is unknown. Unlike AIS, TBI affects all age groups, with the majority of PTCI occurring in those <40 years old [11]. The prevention of AIS after TBI is especially important given the greater lifetime burden of post-TBI disability in younger patients.
2. Pathophysiology
Many subclinical events increase PTCI risk, both at the macroscopic and the cellular level. As a final common pathway, mismatch between metabolic demand and oxygen delivery results in ischemic injury, and if sustained, results in neuronal death. Metabolic demand can be reduced through appropriate sedation and analgesia and by treating hypermetabolic states such as seizures and fever. Oxygen delivery can be optimized by the frequent monitoring of oxygen saturation, partial pressure of blood oxygen, blood pressure, and cerebral perfusion pressure (CPP). Avoidance of hypotension is critical as it exacerbates ischemic injury. Among pre-hospital TBI patients, each 10-point decrease in systolic pressure below the normal range is associated with an 18.8% increase in the adjusted odds of death [14], while the presence of both hypotension and hypoxia predict a significantly higher mortality than either alone [15].
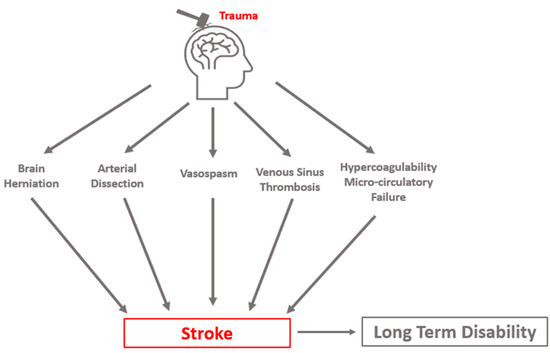
Three distinct clinical syndromes are most causative of clinically relevant PTCI: cerebral herniation, CAD, and post-traumatic vasospasm (PTV, VSP). Stroke may occur in a delayed fashion, highlighting the importance of the early identification and appropriate management of risk factors. Cerebral infarct due to sinus thrombosis can occur but is a less common cause of PTCI. Additional sequelae of TBI, such as coagulopathy and microcirculatory failure, likely exacerbate delayed cerebral ischemia from these mechanisms (Figure 1).
Figure 1. Conceptual schematic of primary etiologies of post-traumatic cerebral infarction.
2.1. Hypercoagulability after Traumatic Brain Injury
Coagulopathy after polytrauma is well described, but also occurs in 36–45% of isolated moderate-to-severe TBI patients [16][17]. As in polytrauma, both hypocoagulable and hypercoagulable states can exist. While hypocoagulability is implicated in hemorrhagic expansion after TBI, hypercoagulability results in an increased risk for PTCI. Proposed causes of hypercoagulability include platelet hyperactivity induced by platelet-activating factor (PAF), increased expression of tissue factor, and early disseminated intravascular coagulation [18][19].
2.2. Brain Herniation
Brain herniation is the most feared complication after TBI, as it is rapidly progressive and potentially fatal. The likelihood of herniation increases with TBI severity. In a recent study of severe TBI, 44% of patients had clinical signs of herniation on presentation [20]. When the clinical exam is limited, herniation might only be appreciated with early neuroimaging performed within 24–48 h of injury [21]. Progression to herniation is associated with mortality rates exceeding 80% [22]. As a predictor of delayed complications and less favorable outcomes, the presence of herniation can have a significant impact on patient management and goals of care.
Brain herniation after TBI can lead to secondary ischemia through several mechanisms. Large territory infarcts are associated with specific herniation syndromes as displaced brain tissue mechanically compresses adjacent vessels. In transtentorial (uncal) herniation, descending supratentorial tissue compresses the posterior cerebral artery (PCA) and its branches, including the calcarine branch, leading to PCA infarction in the inferior temporal and occipital lobe ipsilateral to the herniation. In patients with a reliable clinical exam, a rapid change in level of consciousness and contralateral hemiparesis or posturing is observed. In intubated and sedated patients, pupillary dilation and sluggish reactivity are often the first observable signs [23][24].
2.3. Cervical Artery Dissection
Cervical artery dissection is a relatively uncommon complication of TBI. It is observed in approximately 2% of moderate-to-severe TBI survivors, with carotid and vertebral artery dissections being equally likely [11]. The true prevalence of CAD is likely higher as most patients are asymptomatic; however, when present, it is widely recognized as an important cause of preventable PTCI. Moderate-to-severe TBI survivors with traumatic CAD have a 9.4% incidence of PTCI and are associated with worse functional and cognitive outcomes [11]. The mortality rate from traumatic CAD is difficult to ascertain as patients with severe brain injury often die before a diagnosis is made and likely have multiple factors contributing to death [25].
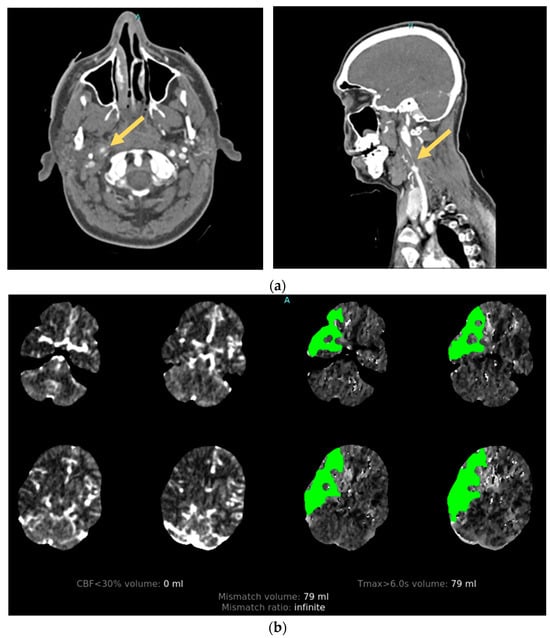
High-impact injuries are strongly associated with CAD. Their development is attributed to rapid acceleration–deceleration leading to hyperextension and rotation of the neck, the stretching of vessels over adjacent vertebral structures, and the production of a tear in the intimal layer of the artery [26]. After endothelial injury, circulating blood under arterial pressure penetrates the damaged vessel wall, separating the intima and media layers. The resulting intramural hematoma can create a combination of macroscopic vascular defects that include vessel occlusion, stenosis, and pseudoaneurysm that can be seen on angiographic imaging [27]. Damage to the arterial intima also exposes subendothelial collagen to the intravascular space, which acts to trigger the intrinsic coagulation pathway, leading to platelet aggregation and activation with thrombi formation [28]. The majority of AIS secondary to CAD is subsequently caused by thromboembolism from the dissection site [29] (Figure 2).
Figure 2. Illustrative case. A patient with traumatic cervical artery dissection after a motor vehicle accident. (a) CT angiogram shows right internal carotid dissection accompanied by pseudoaneurysm formation (arrows). (b) CT perfusion demonstrates a large area of delayed filling in the distribution of the dissected vessel.
2.4. Post-Traumatic Vasospasm
Vasospasm can have a significant impact on functional outcomes after TBI. It is extensively studied in the context of aneurysmal subarachnoid hemorrhage (aSAH), but fewer data are available regarding the epidemiology and pathophysiology of PTV. Although PTV and VSP after aSAH likely share many similarities, a better understanding of the unique characteristics of PTV contributing to secondary brain injury is needed. The consequences of PTV are significant. In an observational study of pediatric patients, a good neurologic outcome (defined as a 30-day GCS score of ≥4) after moderate-to-severe TBI was seen in 76% of patients without VSP, compared to 40% in those with VSP [30].
PTV is not routinely assessed unless clinical signs or symptoms suggest its presence, leading to underdiagnosis. In clinical practice, VSP screening is considered when a focal neurologic deficit or mental status change is not explained by neuroimaging, electroencephalogram (EEG), or comorbid condition. VSP is also considered with new radiographic evidence of ischemia not caused by the primary injury.
2.5. Cerebral Venous Thrombosis
Cerebral venous sinus thrombosis (CVST) can cause stroke by impeding venous flow. Its true incidence after TBI is unknown, but it may be more commonly identified with the increased use of advanced brain and vascular imaging techniques. Although post-TBI CVST often follows a benign course, intracerebral complications such as PTCI, hemorrhage, and cerebral edema can occur and are associated with increased mortality [31][32]. The primary risk factor for acute CVST is skull fracture resulting in sinus injury [33], with CVST rarely occurring without an overlying skull fracture.
2.6. Microthrombosis and Microcirculatory Failure
Ischemic injury burden after TBI may not be grossly appreciated. Microthrombosis, an emerging pathophysiologic concept that occurs in several types of critical illness, is thought to worsen ischemic burden and secondary brain injury in small, distal cerebral vessels. Although the mechanisms are not entirely understood, inflammatory and thrombotic processes appear closely linked [34].
3. Diagnosis
3.1. Clinical Diagnosis of Acute Ischemic Stroke
Post-traumatic cerebral infarction is diagnosed using similar radiographic modalities used in spontaneous AIS, primarily non-contrast computed tomography (CT) and magnetic resonance imaging (MRI) of the brain. Although imaging is essential for anatomic characterization and definitive diagnosis, AIS is first clinically suspected due to either an acute change in neurologic status or failure to neurologically improve. Asymmetry in the motor exam may or may not be apparent. An accurate neurologic examination is often confounded by a low GCS score secondary to several factors commonly present in critically ill patients: traumatic, toxic, and metabolic encephalopathy, use of sedatives and analgesics, and intensive care unit (ICU) delirium. Physical barriers, including behavioral restraints, lines and drains, and bedrest orders also limit comprehensive neurologic testing and should be minimized when safe.
3.2. CT and MRI Neuroimaging Modalities
Current guidelines recommend non-contrast multidetector CT (MDCT) of the brain for the routine initial imaging after TBI [35][36]. MDCT is widely available and can quickly diagnose bony fractures, foreign objects, and acute intracranial hemorrhage (ICH) to identify patients requiring urgent interventions. AIS is identified on MDCT or MRI by the presence of cytotoxic edema in a vascular territory. With MDCT, the sensitivity of detecting AIS is influenced by the duration of ischemia and the size and location of the lesion.
The use of MRI in the initial evaluation of TBI is limited by its availability and longer acquisition times; however, in select patients, its increased sensitivity for ischemia is important in diagnosing PTCI. MRI is often triggered when the neurologic exam or recovery is less than expected based on MDCT or early neuroimaging.
Differentiating PTCI from trauma-related contusion, cerebral edema, surgical complications, or expected post-surgical changes is a diagnostic challenge. The vascular distribution of a brain lesion is helpful and requires a basic understanding of neuroanatomy. The timing and progression of imaged lesions is also useful: contusions are unlikely to become newly apparent several days after TBI, and vasogenic edema from an ICH might be restricted to the perihematomal region.
3.3. Vessel Imaging
3.3.1. Angiography
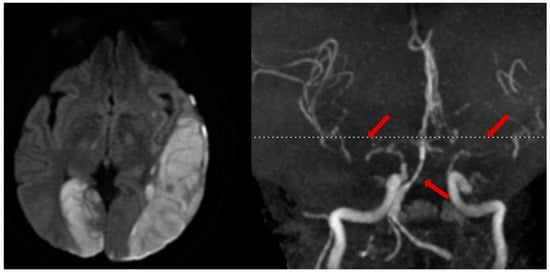
In select cases of suspected or high risk of cervical artery injury, a contrast CT angiogram (CTA) of neck vessels is recommended. A CTA of the head and neck can be rapidly conducted and carries little additional risk. Although a clinical concern over contrast-induced nephropathy exists, the incidence of renal sequelae is low when performed in emergency AIS protocols [37]. Alternatively, an MR angiogram (MRA) with MRI for stroke evaluation can be performed (Figure 3).
Figure 3. Illustrative case. A patient with a traumatic left subdural hematoma after assault, with MRA showing severe vasospasm in the bilateral anterior and middle cerebral arteries, mid-to-distal basilar, and posterior cerebral arteries (right, arrows), leading to acute ischemic strokes in the left MCA and right PCA territories (left).
3.3.2. MR Vessel Wall Imaging: An Emerging Diagnostic Tool
MR vessel wall imaging (VWI) directly visualizes the vessel wall by suppressing blood signals in the artery lumen, allowing for the assessment of vessel wall pathologies that might lead to ischemic infarct. Compared to standard angiographic imaging, MR VWI can differentiate underlying pathologies, accurately evaluate the severity of vessel disease, and monitor its progression. It may be especially useful in evaluating arterial dissection and venous thrombosis. The diagnostic value in arterial dissection has been demonstrated in both cervical and intracranial arteries [38][39][40]. Classic MR VWI presentations of dissection include the presence of a double lumen, “string” sign, or a crescent-shaped intramural hematoma. Venous thrombosis appears as heterogeneous signal intensity within the dark lumen of the venous sinus. MR VWI has shown excellent diagnostic value in acute CVST [41][42], and can be used to predict treatment response to endovascular therapy [43].
3.4. Other Methods of Detecting Ischemic Injury
3.4.1. Transcranial Doppler Ultrasound
Transcranial Doppler utilizes ultrasound-frequency waves to characterize CBF in real time. It can serve as a useful tool to monitor for VSP after neurologic injury. When used in aSAH, a mean flow velocity (MFV) > 120 cm/s in anterior circulation arteries suggests VSP, while a pulsatility index (PI) > 1.2 indicates poor arterial compliance that can suggest distal VSP or cerebral edema. Its use in TBI is less established and requires further clinical study. A recent meta-analysis showed that similar TCD findings (MFV > 120 cm/s, PI > 1.2) are associated with a 9-fold-increased risk of mortality and greater than 3-fold-increased risk for poor clinical outcome after TBI [44]. Pre-hospital and admission TCD can identify patients with cerebral hypoperfusion after TBI [45][46]; however, less is known about its utility in detecting delayed PTCI. Based on experiences in aSAH and spontaneous AIS, TCD can provide early evidence of VSP that may mediate AIS and can also be used to detect the presence of intracranial occlusion and microembolization secondary to proximal cervical artery vasculopathies such as dissection [47][48].
3.4.2. Electroencephalogram
EEG aids in neurodiagnostic assessment by characterizing the electrical activity of the cerebral cortex. The presence of electrographic seizures, epileptiform discharges, and disorganized or slowed waveforms can be indicative of structural lesions such as delayed hemorrhagic expansion or stroke after TBI, but findings may be non-specific. EEG has not been extensively studied in PTCI, but its use in AIS and aSAH suggests a potential role in TBI monitoring. The suppression of all EEG frequencies (termed cortical suppression) has been correlated with a CBF of less than 8–10 mL/100 g/min, and this threshold can be used to detect large vessel stroke [49][50]. Newer continuous EEG modalities, including quantitative EEG, have been proposed as methods for detecting delayed brain ischemia in the ICU [51].
4. Management
4.1. Management of Cerebral Herniation Syndromes
Left untreated, brain herniation leads to irreversible ischemic and mechanical injury and is rapidly fatal. Strategies for early detection and treatment include the implementation of neuromonitoring protocols and the use of stepwise therapy for intracranial hypertension. Early medical treatment is focused on reducing ICP by decreasing intracranial contents including cerebrospinal fluid, cerebral blood volume, and brain water content. The mainstay of medical management for herniation is hyperosmolar therapy with mannitol and hypertonic saline (HTS). Its immediate effect in reversing herniation is targeted at reducing brain water content through the creation of an osmotic gradient that favors water movement from intracellular and interstitial spaces into the vasculature.
There are many practical considerations when using hyperosmolar therapy. Mannitol is commonly used as a first-line therapy, especially outside of a specialized neuroscience ICU, as it is often more readily available and can be easily administered through a peripheral intravenous line. The recommended dose of mannitol is 0.25–1.0 g/kg body weight. Peak effect is seen 15–30 min after infusion, and ICP-lowering effects are expected to last approximately 60 min [52]; however, this is variable in clinical practice. In large or multiple doses, mannitol can cause profound diuresis leading to intravascular volume depletion and hypotension and may not be appropriate in patients in shock or with acute kidney injury [24].
4.2. Management of Cervical Artery Dissection
The rationale for antithrombotic therapy, including antiplatelet and anticoagulation use, is to prevent the embolization of thrombi from the injured artery and to inhibit the occlusion of stenotic vessels. Currently, there is a paucity of high-quality evidence to compare the efficacies of antiplatelet and anticoagulant drugs for stroke prevention in CAD. A prior systematic review of observational studies suggested no benefit to anticoagulation over antiplatelet use [53]. The Cervical Artery Dissection in Stroke Study (CADISS) was the first multicenter, prospective, randomized controlled trial that compared anticoagulation to antiplatelet treatment in acute symptomatic CAD. The study included patients with extracranial carotid or vertebral artery dissection with an onset of symptoms within the past 7 days. Enrollment consisted of 126 patients assigned to antiplatelet treatment and 124 assigned to anticoagulant treatment. Results showed no difference in rates of ipsilateral stroke between the two groups at 3 months (2% [3/126] vs. 1% [1/124], p = 0.63), but the study was underpowered to detect a significant difference in outcome [54]. The study population consisted of mostly spontaneous CAD, which leads to AIS at presumably lower rates than traumatic CAD.
4.3. Management of Traumatic Vasospasm
In the absence of high-quality data, managing PTV poses unique challenges. For patients with aSAH, treatment with calcium-channel blockers (CCBs) reduce morbidity, while components of “Triple-H” therapy (hypervolemia, hypertension, and hemodilution) are often initiated to treat VSP. These interventions, however, could be detrimental in the setting of TBI depending on the severity of injury and associated comorbidities. Induced hypertension and hypervolemia can potentially worsen cerebral edema and ICP if cerebral autoregulation is impaired [55].
4.4. Management of Cerebral Venous Thrombosis
Although therapeutic anticoagulation for a period of 3–6 months is the guideline recommendation for non-traumatic CVST to prevent ischemic complications [56], the presence of intracranial hemorrhage or other non-neurologic traumatic injuries often precludes this. The early use of anticoagulation after hematoma stabilization without bleeding complications has been reported in small case series [57]; however, in the absence of high-quality guidance, treatment is often conservative. The prevention of intravascular volume depletion by maintaining euvolemia is thought to prevent clot propagation. Serial venous imaging can be performed in the acute period to monitor for CVST propagation and change treatment strategies if needed.
References
- Centers for Disease Control and Prevention. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2014; Centers for Disease Control and Prevention, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Frieden, T.R.; Houry, D.; Baldwin, G. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA, USA, 2015.
- Corso, P.; Finkelstein, E.; Miller, T.; Fiebelkorn, I.; Zaloshnja, E. Incidence and lifetime costs of injuries in the United States. Inj. Prev. 2006, 12, 212–218.
- Coronado, V.G.; McGuire, L.C.; Sarmiento, K.; Bell, J.; Lionbarger, M.R.; Jones, C.D.; Geller, A.I.; Khoury, N.; Xu, L. Trends in Traumatic Brain Injury in the U.S. and the public health response: 1995–2009. J. Saf. Res. 2012, 43, 299–307.
- Hinson, H.E.; Rowell, S.; Schreiber, M. Clinical evidence of inflammation driving secondary brain injury: A systematic review. J. Trauma Inj. Infect. Crit. Care 2015, 78, 184–191.
- Pilitsis, J.G.; Coplin, W.M.; O’Regan, M.H.; Wellwood, J.M.; Diaz, F.G.; Fairfax, M.R.; Michael, D.B.; Phillis, J.W. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci. Lett. 2003, 349, 136–138.
- Engel, S.; Schluesener, H.; Mittelbronn, M.; Seid, K.; Adjodah, D.; Wehner, H.D. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 2000, 100, 313–322.
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98.
- Poblete, R.A.; Arenas, M.; Sanossian, N.; Freeman, W.D.; Louie, S.G. The role of bioactive lipids in attenuating the neuroinflammatory cascade in traumatic brain injury. Ann. Clin. Transl. Neurol. 2020, 7, 2524–2534.
- Beaumont, A.; Marmarou, A.; Hayasaki, K.; Barzo, P.; Fatouros, P.; Corwin, F.; Marmarou, C.; Dunbar, J. The permissive nature of blood brain barrier (BBB) opening in edema formation following traumatic brain injury. Acta Neurochir. Suppl. 2000, 76, 125–129.
- Kowalski, R.G.; Haarbauer-Krupa, J.K.; Bell, J.M.; Corrigan, J.D.; Hammond, F.M.; Torbey, M.T.; Hofmann, M.C.; Dams-O’Connor, K.; Miller, A.C.; Whiteneck, G.G. Acute Ischemic Stroke after Moderate to Severe Traumatic Brain Injury: Incidence and Impact on Outcome. Stroke 2017, 48, 1802–1809.
- Bae, D.H.; Choi, K.S.; Yi, H.J.; Chun, H.J.; Ko, Y.; Bak, K.H. Cerebral Infarction after Traumatic Brain Injury: Incidence and Risk Factors. Korean J. Neurotrauma 2014, 10, 35–40.
- Tian, H.L.; Geng, Z.; Cui, Y.H.; Hu, J.; Xu, T.; Cao, H.; Chen, S.; Chen, H. Risk factors for posttraumatic cerebral infarction in patients with moderate or severe head trauma. Neurosurg. Rev. 2008, 31, 431–437, discussion 436–437.
- Spaite, D.W.; Hu, C.; Bobrow, B.J.; Chikani, V.; Sherrill, D.; Barnhart, B.; Gaither, J.B.; Denninghoff, K.R.; Viscusi, C.; Mullins, T.; et al. Mortality and Prehospital Blood Pressure in Patients With Major Traumatic Brain Injury: Implications for the Hypotension Threshold. JAMA Surg. 2017, 152, 360–368.
- Spaite, D.W.; Hu, C.; Bobrow, B.J.; Chikani, V.; Barnhart, B.; Gaither, J.B.; Denninghoff, K.R.; Adelson, P.D.; Keim, S.M.; Viscusi, C.; et al. The Effect of Combined Out-of-Hospital Hypotension and Hypoxia on Mortality in Major Traumatic Brain Injury. Ann. Emerg. Med. 2016, 69, 62–72.
- Talving, P.; Benfield, R.; Hadjizacharia, P.; Inaba, K.; Chan, L.S.; Demetriades, D. Coagulopathy in severe traumatic brain injury: A prospective study. J. Trauma Inj. Infect. Crit. Care 2009, 66, 55–62, discussion 61–62.
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Inaba, K.; Lam, L.; Plurad, D.; Demetriades, D. Time course of coagulopathy in isolated severe traumatic brain injury. Injury 2010, 41, 924–928.
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J.F. Traumatic brain injury-associated coagulopathy. J. Neurotrauma 2012, 29, 2597–2605.
- Laroche, M.; Kutcher, M.E.; Huang, M.C.; Cohen, M.J.; Manley, G.T. Coagulopathy after traumatic brain injury. Neurosurgery 2012, 70, 1334–1345.
- Chestnut, R.; Ghajar, J.; Maas, A.I.R.; Marion, D.W.; Servadei, F.; Teasdale, G.M.; Unterberg, A.; von Holst, H.; Walters, B.C. Early indicators of prognosis in severe traumatic brain injury: Brain Trauma Foundation. J. Neurotrauma 2000, 57, 1–103.
- Currie, S.; Saleem, N.; Straiton, J.A.; Macmullen-Price, J.; Warren, D.J.; Craven, I.J. Imaging assessment of traumatic brain injury. Postgrad. Med. J. 2015, 92, 41–50.
- Lan, Z.; Richard, S.A.; Li, Q.; Wu, C.; Zhang, Q.; Chen, R.; Yang, C. Outcomes of patients undergoing craniotomy and decompressive craniectomy for severe traumatic brain injury with brain herniation: A retrospective study. Medicine 2020, 99, e22742.
- Ropper, A.H. Herniation. Handb. Clin. Neurol. 2008, 90, 79–98.
- Sundstrøm, T.; Grände, P.; Juul, N.; Kock-Jensen, C.; Romner, B.; Wester, K. Management of Severe Traumatic Brain Injury Evidence, Tricks, and Pitfalls; Springer: Berlin/Heidelberg, Germany, 2012.
- Debette, S.; Leys, D. Cervical-artery dissections: Predisposing factors, diagnosis, and outcome. Lancet Neurol. 2009, 8, 668–678.
- Kraus, R.R.; Bergstein, J.M.; DeBord, J.R. Diagnosis, treatment, and outcome of blunt carotid arterial injuries. Am. J. Surg. 1999, 178, 190–193.
- Singh, R.R.; Barry, M.C.; Ireland, A.; Bouchier Hayes, D. Current diagnosis and management of blunt internal carotid artery injury. Eur. J. Vasc. Endovasc. Surg. 2004, 27, 577–584.
- Baumgartner, R.W.; Arnold, M.; Baumgartner, I.; Mosso, M.; Gönner, F.; Studer, A.; Schroth, G.; Schuknecht, B.; Sturzenegger, M. Carotid dissection with and without ischemic events: Local symptoms and cerebral artery findings. Neurology 2001, 57, 827–832.
- Morel, A.; Naggar, O.; Touzé, E.; Raymond, J.; Mas, J.; Meder, J.; Oppenheim, C. Mechanism of ischemic infarct in spontaneous cervical artery dissection. Stroke 2012, 43, 1354–1361.
- O’Brien, N.F.; Reuter-Rice, K.E.; Khanna, S.; Peterson, B.M.; Quinto, K.B. Vasospasm in children with traumatic brain injury. Intensiv. Care Med. 2010, 36, 680–687.
- Netteland, D.F.; Mejlaender-Evjensvold, M.; Skaga, N.O.; Sandset, E.C.; Aarhus, M.; Helseth, E. Cerebral venous thrombosis in traumatic brain injury: A cause of secondary insults and added mortality. J. Neurosurg. 2021, 134, 1912–1920.
- Qureshi, A.I.; Sahito, S.; Liaqat, J.; Chandrasekaran, P.N.; Siddiq, F. Traumatic Injury of Major Cerebral Venous Sinuses Associated with Traumatic Brain Injury or Head and Neck Trauma: Analysis of National Trauma Data Bank. J. Vasc. Interv. Neurol. 2020, 11, 27–33.
- Delgado Almandoz, J.E.; Kelly, H.R.; Schaefer, P.W.; Lev, M.H.; Gonzalez, R.G.; Romero, J.M. Prevalence of traumatic dural venous sinus thrombosis in high-risk acute blunt head trauma patients evaluated with multidetector CT venography. Radiology 2010, 255, 570–577.
- Ekdahl, K.N.; Teramura, Y.; Hamad, O.A.; Asif, S.; Duehrkop, C.; Fromell, K.; Gustafson, E.; Hong, J.; Kozarcanin, H.; Magnusson, P.U.; et al. Dangerous liaisons: Complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 2016, 274, 245–269.
- Shetty, V.S.; Reis, M.N.; Aulino, J.M.; Berger, K.L.; Broder, J.; Choudhri, A.F.; Kendi, A.T.; Kessler, M.M.; Kirsch, C.F.; Luttrull, M.D.; et al. ACR Appropriateness Criteria Head Trauma. J. Am. Coll. Radiol. 2016, 13, 668–679.
- Ryan, M.E.; Palasis, S.; Saigal, G.; Singer, A.D.; Karmazyn, B.; Dempsey, M.E.; Dillman, J.R.; Dory, C.E.; Garber, M.; Hayes, L.L.; et al. ACR Appropriateness Criteria head trauma--child. J. Am. Coll. Radiol. 2014, 11, 939–947.
- Hopyan, J.J.; Gladstone, D.J.; Mallia, G.; Schiff, J.; Fox, A.J.; Symons, S.P.; Buck, B.H.; Black, S.E.; Aviv, R.I. Renal safety of CT angiography and perfusion imaging in the emergency evaluation of acute stroke. Am. J. Neuroradiol. 2008, 29, 1826–1830.
- Yuan, X.; Cui, X.; Gu, H.; Wang, M.; Dong, Y.; Cai, S.; Feng, X.; Wang, X. Evaluating cervical artery dissections in young adults: A comparison study between high-resolution MRI and CT angiography. Int. J. Cardiovasc. Imaging 2020, 36, 1113–1119.
- Zhu, X.; Shan, Y.; Guo, R.; Zheng, T.; Zhang, X.; Liu, Z.; Liu, K. Three-Dimensional High-Resolution Magnetic Resonance Imaging for the Assessment of Cervical Artery Dissection. Front. Aging Neurosci. 2022, 14, 785661.
- Chen, S.; Ma, G.; Zhang, P.; Kang, Q. Isolated traumatic supraclinoid internal carotid artery dissection diagnosed by high-resolution vessel wall MRI. Br. J. Neurosurg. 2021, 37, 1801–1804.
- Yang, X.; Wu, F.; Liu, Y.; Duan, J.; Fisher, M.; Ji, X.; Meng, R.; Zhang, H.; Fan, Z.; Yang, Q. Diagnostic performance of MR black-blood thrombus imaging for cerebral venous thrombosis in real-world clinical practice. Eur. Radiol. 2021, 32, 2041–2049.
- Hakim, A.; Kurmann, C.; Pospieszny, K.; Meinel, T.R.; Shahin, M.A.; Heldner, M.R.; Umarova, R.; Jung, S.; Arnold, M.; El-Koussy, M. Diagnostic Accuracy of High-Resolution 3D T2-SPACE in Detecting Cerebral Venous Sinus Thrombosis. AJNR Am. J. Neuroradiol. 2022, 43, 881–886.
- Yang, X.; Wu, F.; Liu, Y.; Duan, J.; Meng, R.; Chen, J.; Li, D.; Fan, Z.; Fisher, M.; Yang, Q.; et al. Predictors of successful endovascular treatment in severe cerebral venous sinus thrombosis. Ann. Clin. Transl. Neurol. 2019, 6, 755–761.
- Fatima, N.; Shuaib, A.; Chughtai, T.S.; Ayyad, A.; Saqqur, M. The Role of Transcranial Doppler in Traumatic Brain Injury: A Systemic Review and Meta-Analysis. Asian J. Neurosurg. 2019, 14, 626–633.
- Ract, C.; Le Moigno, S.; Bruder, N.; Vigué, B. Transcranial Doppler ultrasound goal-directed therapy for the early management of severe traumatic brain injury. Intensiv. Care Med. 2007, 33, 645–651.
- Tazarourte, K.; Atchabahian, A.; Tourtier, J.P.; David, J.; Ract, C.; Savary, D.; Monchi, M.; Vigué, B. Pre-hospital transcranial Doppler in severe traumatic brain injury: A pilot study. Acta Anaesthesiol. Scand. 2011, 55, 422–428.
- Tsivgoulis, G.; Sharma, V.K.; Lao, A.Y.; Malkoff, M.D.; Alexandrov, A.V. Validation of transcranial Doppler with computed tomography angiography in acute cerebral ischemia. Stroke 2007, 38, 1245–1249.
- Demchuk, A.M.; Christou, I.; Wein, T.H.; Felberg, R.A.; Malkoff, M.; Grotta, J.C.; Alexandrov, A.V. Accuracy and criteria for localizing arterial occlusion with transcranial Doppler. J. Neuroimaging 2000, 10, 1–12.
- Bhattarai, S.; Zhang, X.; Tuerxun, T. EEG and SPECT changes in acute ischemic stroke. J. Neurol. Neurophysiol. 2014, 5, 2.
- Schneider, A.L.; Jordan, K.G. Regional attenuation without delta (RAWOD): A distinctive EEG pattern that can aid in the diagnosis and management of severe acute ischemic stroke. Am. J. Electroneurodiagnostic Technol. 2005, 45, 102–117.
- Foreman, B.; Claassen, J. Quantitative EEG for the detection of brain ischemia. Crit. Care 2012, 16, 216.
- Kølsen-Petersen, J.A. Osmotherapy. In Management of Severe Traumatic Brain Injury: Evidence, Tricks, and Pitfalls; Sundstrom, T., Grände, P., Juul, N., Kock-Jensen, C., Romner, B., Wester, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 293–302.
- Lyrer, P.; Engelter, S. Antithrombotic drugs for carotid artery dissection. Stroke 2004, 35, 613–614.
- CADISS trial investigators. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): A randomised trial. Lancet Neurol. 2015, 14, 361–367.
- Lee, K.H.; Lukovits, T.; Friedman, J.A. “Triple-H” therapy for cerebral vasospasm following subarachnoid hemorrhage. Neurocritical Care 2006, 4, 68–76.
- Saposnik, G.; Barinagarrementeria, F.; Brown, R.D., Jr.; Bushnell, C.D.; Cucchiara, B.; Cushman, M.; deVeber, G.; Ferro, J.M.; Tsai, F.Y. Diagnosis and management of cerebral venous thrombosis: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 1158–1192.
- Grangeon, L.; Gilard, V.; Ozkul-Wermester, O.; Lefaucheur, R.; Curey, S.; Gerardin, E.; Derrey, S.; Maltete, D.; Magne, N.; Triquenot, A. Management and outcome of cerebral venous thrombosis after head trauma: A case series. Rev. Neurol. 2017, 173, 411–417.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
665
Revisions:
2 times
(View History)
Update Date:
20 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No