 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abdulhameed Al-Ghabkari | -- | 3681 | 2024-02-19 14:52:41 | | | |
| 2 | Sirius Huang | Meta information modification | 3681 | 2024-02-21 02:05:57 | | |
Video Upload Options
Despite therapeutic advances, the treatment of brain tumors, including glioblastoma (GBM), an aggressive primary brain tumor associated with poor prognosis and resistance to therapy, remains a significant challenge. Receptor tyrosine kinases (RTKs) are critical during development and in adulthood. Dysregulation of RTKs through activating mutations and gene amplification contributes to many human cancers and provides attractive therapeutic targets for treatment. Under physiological conditions, the Met RTK, the hepatocyte growth factor/scatter factor (HGF/SF) receptor, promotes fundamental signaling cascades that modulate epithelial-to-mesenchymal transition (EMT) involved in tissue repair and embryogenesis. In cancer, increased Met activity promotes tumor growth and metastasis by providing signals for proliferation, survival, and migration/invasion. Recent clinical genomic studies have unveiled multiple mechanisms by which MET is genetically altered in GBM, including focal amplification, chromosomal rearrangements generating gene fusions, and a splicing variant mutation (exon 14 skipping, METex14del). Notably, MET overexpression contributes to chemotherapy resistance in GBM by promoting the survival of cancer stem-like cells. This is linked to distinctive Met-induced pathways, such as the upregulation of DNA repair mechanisms, which can protect tumor cells from the cytotoxic effects of chemotherapy.
1. Introduction
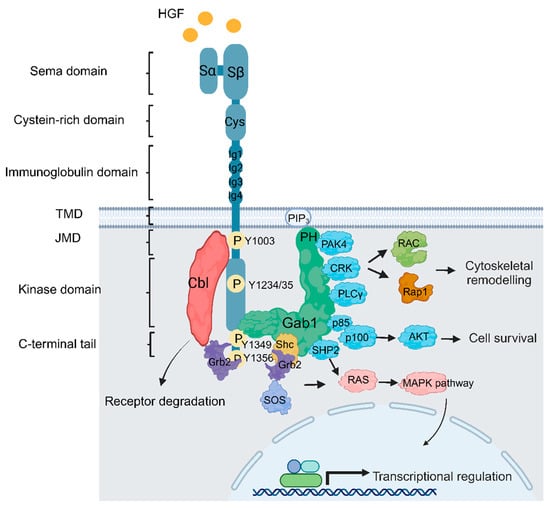
2. Met Structure and Function
3. MET/HGF Dysregulation and Oncogenic Paradigms in GBM
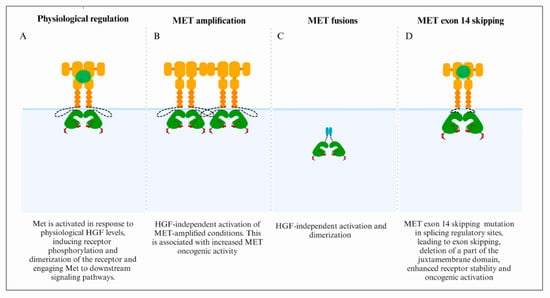
3.1. MET Focal Amplification
3.2. Fusion Genes
3.3. MET Exon 14 Skipping
References
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406.
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165.
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142.
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43.
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848.
- Wu, F.; Chai, R.C.; Wang, Z.; Liu, Y.Q.; Zhao, Z.; Li, G.Z.; Jiang, H.Y. Molecular classification of IDH-mutant glioblastomas based on gene expression profiles. Carcinogenesis 2019, 40, 853–860.
- Bausart, M.; Preat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35.
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell. Mol. Immunol. 2018, 15, 737–739.
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189.
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401.
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798.
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812.
- Skiriute, D.; Vaitkiene, P.; Saferis, V.; Asmoniene, V.; Skauminas, K.; Deltuva, V.P.; Tamasauskas, A. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012, 12, 218.
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251.
- Wintheiser, G.A.; Silberstein, P. Physiology, Tyrosine Kinase Receptors. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Stern, Y.E.; Al-Ghabkari, A.; Monast, A.; Fiset, B.; Aboualizadeh, F.; Yao, Z.; Stagljar, I.; Walsh, L.A.; Duhamel, S.; Park, M. Met-HER3 crosstalk supports proliferation via MPZL3 in MET-amplified cancer cells. Cell. Mol. Life Sci. 2022, 79, 178.
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270.
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290.
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456.
- Eckerich, C.; Zapf, S.; Fillbrandt, R.; Loges, S.; Westphal, M.; Lamszus, K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int. J. Cancer 2007, 121, 276–283.
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cazares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956.
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33.
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904.
- Rodrigues, G.A.; Park, M. Dimerization mediated through a leucine zipper activates the oncogenic potential of the met receptor tyrosine kinase. Mol. Cell Biol. 1993, 13, 6711–6722.
- Dean, M.; Park, M.; Vande Woude, G.F. Characterization of the rearranged tpr-met oncogene breakpoint. Mol. Cell Biol. 1987, 7, 921–924.
- Cai, K.; Zhang, X.; Bai, X.C. Cryo-electron Microscopic Analysis of Single-Pass Transmembrane Receptors. Chem. Rev. 2022, 122, 13952–13988.
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. The Hepatocyte Growth Factor Receptor: Structure, Function and Pharmacological Targeting in Cancer. Curr. Signal Transduct. Ther. 2011, 6, 146–151.
- Maulik, G.; Shrikhande, A.; Kijima, T.; Ma, P.C.; Morrison, P.T.; Salgia, R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002, 13, 41–59.
- Sattler, M.; Ma, P.C.; Salgia, R. Therapeutic targeting of the receptor tyrosine kinase Met. Cancer Treat. Res. 2004, 119, 121–138.
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305.
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor: Molecular structure, roles in liver regeneration, and other biological functions. Crit. Rev. Oncog. 1992, 3, 27–54.
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702.
- Toshikazu, N. Structure and function of hepatocyte growth factor. Prog. Growth Factor Res. 1991, 3, 67–85.
- Uchikawa, E.; Chen, Z.; Xiao, G.-Y.; Zhang, X.; Bai, X.-C. Structural basis of the activation of c-MET receptor. Nat. Commun. 2021, 12, 4074.
- Frigault, M.M.; Naujokas, M.A.; Park, M. Gab2 requires membrane targeting and the Met binding motif to promote lamellipodia, cell scatter, and epithelial morphogenesis downstream from the Met receptor. J. Cell Physiol. 2008, 214, 694–705.
- Lock, L.S.; Maroun, C.R.; Naujokas, M.A.; Park, M. Distinct recruitment and function of Gab1 and Gab2 in Met receptor-mediated epithelial morphogenesis. Mol. Biol. Cell 2002, 13, 2132–2146.
- Mood, K.; Saucier, C.; Bong, Y.S.; Lee, H.S.; Park, M.; Daar, I.O. Gab1 is required for cell cycle transition, cell proliferation, and transformation induced by an oncogenic met receptor. Mol. Biol. Cell 2006, 17, 3717–3728.
- Lock, L.S.; Frigault, M.M.; Saucier, C.; Park, M. Grb2-independent recruitment of Gab1 requires the C-terminal lobe and structural integrity of the Met receptor kinase domain. J. Biol. Chem. 2003, 278, 30083–30090.
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Mol. Biol. Cell 2007, 18, 76–83.
- Maroun, C.R.; Holgado-Madruga, M.; Royal, I.; Naujokas, M.A.; Fournier, T.M.; Wong, A.J.; Park, M. The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell Biol. 1999, 19, 1784–1799.
- Rajadurai, C.V.; Havrylov, S.; Zaoui, K.; Vaillancourt, R.; Stuible, M.; Naujokas, M.; Zuo, D.; Tremblay, M.L.; Park, M. Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J. Cell Sci. 2012, 125, 2940–2953.
- Lai, A.Z.; Abella, J.V.; Park, M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009, 19, 542–551.
- Chen, S.Y.; Chen, H.C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell Biol. 2006, 26, 5155–5167.
- McCall-Culbreath, K.D.; Li, Z.; Zutter, M.M. Crosstalk between the alpha2beta1 integrin and c-met/HGF-R regulates innate immunity. Blood 2008, 111, 3562–3570.
- Yamamoto, N.; Mammadova, G.; Song, R.X.; Fukami, Y.; Sato, K. Tyrosine phosphorylation of p145met mediated by EGFR and Src is required for serum-independent survival of human bladder carcinoma cells. J. Cell Sci. 2006, 119, 4623–4633.
- Khoury, H.; Naujokas, M.A.; Zuo, D.; Sangwan, V.; Frigault, M.M.; Petkiewicz, S.; Dankort, D.L.; Muller, W.J.; Park, M. HGF converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol. Biol. Cell 2005, 16, 550–561.
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043.
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin-like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther. 2006, 5, 1676–1682.
- Salokas, K.; Liu, X.; Ohman, T.; Chowdhury, I.; Gawriyski, L.; Keskitalo, S.; Varjosalo, M. Physical and functional interactome atlas of human receptor tyrosine kinases. EMBO Rep. 2022, 23, e54041.
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87.
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469.
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520.
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104.
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658.
- Jonsson, P.; Lin, A.L.; Young, R.J.; DiStefano, N.M.; Hyman, D.M.; Li, B.T.; Berger, M.F.; Zehir, A.; Ladanyi, M.; Solit, D.B.; et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin. Cancer Res. 2019, 25, 5537–5547.
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120.
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029.
- Deshpande, V.; Luebeck, J.; Nguyen, N.D.; Bakhtiari, M.; Turner, K.M.; Schwab, R.; Carter, H.; Mischel, P.S.; Bafna, V. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 2019, 10, 392.
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148.
- Petterson, S.A.; Dahlrot, R.H.; Hermansen, S.K.; KA Munthe, S.; Gundesen, M.T.; Wohlleben, H.; Rasmussen, T.; Beier, C.P.; Hansen, S.; Kristensen, B.W. High levels of c-Met is associated with poor prognosis in glioblastoma. J. Neuro-Oncol. 2015, 122, 517–527.
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin. Cancer Res. 2005, 11, 4479–4486.
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33.
- Joo, K.M.; Jin, J.; Kim, E.; Ho Kim, K.; Kim, Y.; Gu Kang, B.; Kang, Y.J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838.
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med. 2016, 8, 550–568.
- Lolkema, M.P.; Bohets, H.H.; Arkenau, H.T.; Lampo, A.; Barale, E.; de Jonge, M.J.A.; van Doorn, L.; Hellemans, P.; de Bono, J.S.; Eskens, F. The c-Met Tyrosine Kinase Inhibitor JNJ-38877605 Causes Renal Toxicity through Species-Specific Insoluble Metabolite Formation. Clin. Cancer Res. 2015, 21, 2297–2304.
- Sung, V.Y.C.; Knight, J.F.; Johnson, R.M.; Stern, Y.E.; Saleh, S.M.; Savage, P.; Monast, A.; Zuo, D.; Duhamel, S.; Park, M. Co-dependency for MET and FGFR1 in basal triple-negative breast cancers. NPJ Breast Cancer 2021, 7, 36.
- Kim, P.L. Targeting gene fusions in glioma. Curr. Opin. Neurol. 2021, 34, 840–847.
- Charest, A.; Lane, K.; McMahon, K.; Park, J.; Preisinger, E.; Conroy, H.; Housman, D. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003, 37, 58–71.
- You, G.; Fan, X.; Hu, H.; Jiang, T.; Chen, C.C. Fusion Genes Altered in Adult Malignant Gliomas. Front. Neurol. 2021, 12, 715206.
- Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320.
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773.
- Huang, R.; Liu, Y.; Wang, K.; Wang, Z.; Zhang, C.; Zhang, W.; Zhao, Z.; Li, G.; Huang, L.; Chang, Y.; et al. High-sensitive clinical diagnostic method for PTPRZ1-MET and the characteristic protein structure contributing to ligand-independent MET activation. CNS Neurosci. Ther. 2021, 27, 617–628.
- Lai, A.Z.; Cory, S.; Zhao, H.; Gigoux, M.; Monast, A.; Guiot, M.C.; Huang, S.; Tofigh, A.; Thompson, C.; Naujokas, M.; et al. Dynamic reprogramming of signaling upon met inhibition reveals a mechanism of drug resistance in gastric cancer. Sci. Signal 2014, 7, ra38.
- Zeng, A.L.; Yan, W.; Liu, Y.W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381.
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e1618.
- Lamorte, L.; Park, M. The receptor tyrosine kinases: Role in cancer progression. Surg. Oncol. Clin. N. Am. 2001, 10, 271–288.
- Peschard, P.; Park, M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 2007, 26, 1276–1285.
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.-L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6.
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004.
- Peschard, P.; Park, M. Escape from Cbl-mediated downregulation: A recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 2003, 3, 519–523.
- Hammond, D.E.; Urbé, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770.
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859.
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289.
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges. Lung Cancer 2021, 12, 35–50.
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, PO.20.00516.
- Recondo, G.; Che, J.; Jänne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934.
- Lee, C.C.; Yamada, K.M. Identification of a novel type of alternative splicing of a tyrosine kinase receptor. Juxtamembrane deletion of the c-met protein kinase C serine phosphorylation regulatory site. J. Biol. Chem. 1994, 269, 19457–19461.
- Lee, J.; Ou, S.H.I.; Lee, J.M.; Kim, H.C.; Hong, M.; Kim, S.Y.; Jang, J.; Ahn, S.; Kang, S.Y.; Lee, S.; et al. Gastrointestinal malignancies harbor actionable MET exon 14 deletions. Oncotarget 2015, 6, 28211–28222.
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022.
- Sohn, S.-H.; Sul, H.J.; Kim, B.J.; Zang, D.Y. Responses to the Tepotinib in Gastric Cancers with MET Amplification or MET Exon 14 Skipping Mutations and High Expression of Both PD-L1 and CD44. Cancers 2022, 14, 3444.
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Si-Yang, L.; Lan-Ying, G.; An-Na, L.; Na-Na, L.; Hong-Fei, G.; Jian, S.; Jin-Ji, Y.; Xu-Chao, Z.; Yang, S.; Zhong-Yi, D.; et al. The Unique Characteristics of MET Exon 14 Mutation in Chinese Patients with NSCLC. J. Thorac. Oncol. 2016, 11, 1503–1510.
- Clotilde, D.; Frédéric, L.; Fabienne, E.; Zoulika, K.; Martin, F.; Shéhérazade, S.; Simon, B.; Valérie, G.; Philippe, J.; Marie-Christine, C.; et al. Optimization of Routine Testing for MET Exon 14 Splice Site Mutations in NSCLC Patients. J. Thorac. Oncol. 2018, 13, 1873–1883.
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180.
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.-W.; Hida, T.; De Jonge, M.J.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol. 2019, 37, 9004.
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943.
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J. Thorac. Oncol. 2017, 12, 152–156.
- Cierra, H.; Kristen, A.B.; Garland, A.; Luis, V.; Daniel, J.G.; Tian, Z. Control of renal cell carcinoma brain metastases with cabozantinib following progression on immune checkpoint inhibitor therapy. Curr. Probl. Cancer Case Rep. 2021, 3, 100060.

