Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Spiros Vlahopoulos | -- | 3011 | 2024-02-19 14:50:55 | | | |
| 2 | Peter Tang | Meta information modification | 3011 | 2024-02-20 02:33:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Varisli, L.; Vlahopoulos, S. Epithelial–Mesenchymal Transition in Acute Leukemias. Encyclopedia. Available online: https://encyclopedia.pub/entry/55183 (accessed on 24 June 2026).
Varisli L, Vlahopoulos S. Epithelial–Mesenchymal Transition in Acute Leukemias. Encyclopedia. Available at: https://encyclopedia.pub/entry/55183. Accessed June 24, 2026.
Varisli, Lokman, Spiros Vlahopoulos. "Epithelial–Mesenchymal Transition in Acute Leukemias" Encyclopedia, https://encyclopedia.pub/entry/55183 (accessed June 24, 2026).
Varisli, L., & Vlahopoulos, S. (2024, February 19). Epithelial–Mesenchymal Transition in Acute Leukemias. In Encyclopedia. https://encyclopedia.pub/entry/55183
Varisli, Lokman and Spiros Vlahopoulos. "Epithelial–Mesenchymal Transition in Acute Leukemias." Encyclopedia. Web. 19 February, 2024.
Copy Citation
Epithelial–mesenchymal transition (EMT) is a metabolic process that confers phenotypic flexibility to cells and the ability to adapt to new functions. This transition is critical during embryogenesis and is required for the differentiation of many tissues and organs. EMT can also be induced in advanced-stage cancers, leading to further malignant behavior and chemotherapy resistance, resulting in an unfavorable prognosis for patients.
EMT
leukemia
neoplasia
cell adhesion
signaling
1. Introduction
Transitions between epithelial and mesenchymal phenotypes have been studied for decades, and characterized by their dynamic nature, which enables cells to adapt to new functions, according to the needs of the organism [1]. Epithelial cells that undergo epithelial–mesenchymal transition (EMT) lose their junctions and baso-apical polarity, reprogram their metabolism, and acquire a back-to-front polarity that confers them the ability to migrate and invade surrounding tissues. EMT enables the formation of the body plan and the differentiation of multiple tissues and organs. In a mature organism, EMT contributes to tissue repair. In pathology, EMT is involved in organ fibrosis and in cancer progression; in the latter, cells acquire developmental plasticity, migratory and invasive properties, and resistance to apoptosis, senescence, and destruction by the immune system [2].
EMT is activated by various dynamic stimuli from the local microenvironment, including growth factors and cytokines, hypoxia, and contact with the surrounding extracellular matrix (ECM); the mechanism entails specific switching of gene expression programs, which are initiated by transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP), Wnt-β-catenin, Notch, Hedgehog, and receptor tyrosine kinases [3]. These switching programs in turn activate sequence-specific transcription factors (TFs) to turn on the expression of downstream genes [4]. These downstream genes are recognized as the hallmarks of EMT manifestation and encode structural proteins and cell adhesion molecules such as vimentin, N-cadherin, fibronectins, smooth muscle actin, as well as matrix metalloproteases [5]. It must be noted that TGF-β modulates inflammatory processes and has a pronounced impact on tissue homeostasis [6].
In the mammalian fetus, EMT stromal cells of the hepatic portal triads produce fibronectin, which is bound by late-stage erythroid cells to regulate their differentiation; EMT stromal cells transform the microenvironment to support the emergence, expansion, and maintenance of fetal hematopoietic development during the mid-gestational stage [7].
In the normal bone marrow (BM), hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity are controlled by TFs ZEB1 and ZEB2, which operate EMT signaling pathways [8][9]. Knocking out Zeb2 in the BM promotes a phenotype with several features that resemble human myeloproliferative disorders, such as BM fibrosis, splenomegaly, and extramedullary hematopoiesis [10]. After colonizing the fetal liver, Zeb2-deficient hematopoietic stem/progenitor cells (HSPCs) exhibit altered adhesion and homing properties, and fail to reenter the blood circulation to colonize the BM cavity [8].
2. Variations of the EMT Theme in Cancer
In cancer, the activation of EMT switching mechanisms enables phenomena that are linked to de-differentiation and migration, which in combination with the reverse process from EMT, namely MET (mesenchymal to epithelial transition), facilitates metastasis [11][12][13][14][15][16]. What is important is that EMT-like phenomena confer a substantial degree of dynamic plasticity to cancer cell clones, which can function as cancer stem cells that have the properties of drug resistance and tumor initiation [17]. It must be noted that EMT and cancer stem cells (CSCs) do not represent a fixed state of phenotype, but reflect a dynamic flux of adaptive biological responses of malignant cells to drug treatment, oxidant stress, and metabolite alterations in their microenvironment; a characterized feature of CSCs, namely increased expression of aldehyde dehydrogenase (ALDH) enzymes, is linked to radiation resistance and tumor recurrence [17][18]. In malignancy, ALDH enzyme expression is not fixed in a specific cell type but fluctuates according to the disease state, stromal niche, and other factors, and is most likely involved in mediating metabolic adaptation and at least part of the CSCs’ resistance to drug-induced oxidative stress [17][19][20][21][22][23]. ALDH expression in hybrid EMT-like tumor stages has been reported, with example CSCs from ovarian clear cell carcinoma [24].
3. Maintaining and Keeping of Hematopoietic Stem Cells (HSCs) in the BM
All blood cells, including immune system cells, develop from HSCs in the BM [25]. The BM is a highly complex environment composed of many hematopoietic and non-hematopoietic cells and non-cellular components (Figure 1) [26]. The HSCs are not randomly distributed in the BM and reside in specific microenvironments called niches [27]. BM niches contain a highly complex set of non-cellular factors, including various cytokines and growth factors, that are critical for regulating the functions of HSCs in the niche. Although multiple non-cellular factors have been implicated in the regulation of HSCs in the BM niche, the TGF-β family of signaling molecules plays a special role in this regulation. TGF-β is abundant in the BM milieu and is mainly produced and secreted by both hematopoietic cells and various BM niche cells such as megakaryocytes and non-myelinating Schwann cells [28][29][30]. TGF-β has been shown to promote HSC quiescence in BM and to strongly inhibit HSC growth. Neutralization of TGF-β by monoclonal antibody results in an increase in early progenitor cells from quiescent HSCs [31]. In addition, in vivo depletion of megakaryocytes was shown to promote differentiation and to inhibit quiescence in HSCs [32]. However, potential oncogenic functions of TGF-β have also been reported. For example, hypoxic BM microenvironment-dependent stimulation of TGF-β signaling induces CXCR4 expression and, thereby, promotes the survival of chemoresistant LSCs [33]. Consistent with this, it has been shown that inhibition of TGF-β and CXCR4 results in improved survival in an fms-related receptor tyrosine kinase 3 (flt3)-mutated acute myeloid leukemia (AML) model [33]. TGF-β in the BM milieu is also involved in the regulation of osteoblast differentiation from MSCs, but it should be noted that TGF-β signaling has opposite effects in the early and late stages of chondrogenic differentiation [34]. Indeed, TGF-β signaling is associated with multiple intracellular signaling mechanisms, including SMAD, MAPK, and AKT, and TGF-β-regulated chondrogenesis in BM is associated with crosstalk between MAPK, Wnt, and N-cadherin signaling [35]. Furthermore, the inhibition of BMP signaling by the deletion of the gene that encodes bone morphogenetic protein receptor type 1A (bmpr1a) in HSCs and stromal cells increased N-cadherin expression in both osteoblasts and HSCs [36]. The interactions between HSCs and various cellular components of the niche regulate the characteristics of HSCs, including self-renewal and quiescence, which are crucial for maintaining and sustaining the HSC pool [37], and these interactions are mainly provided by adhesion molecules [38][39].
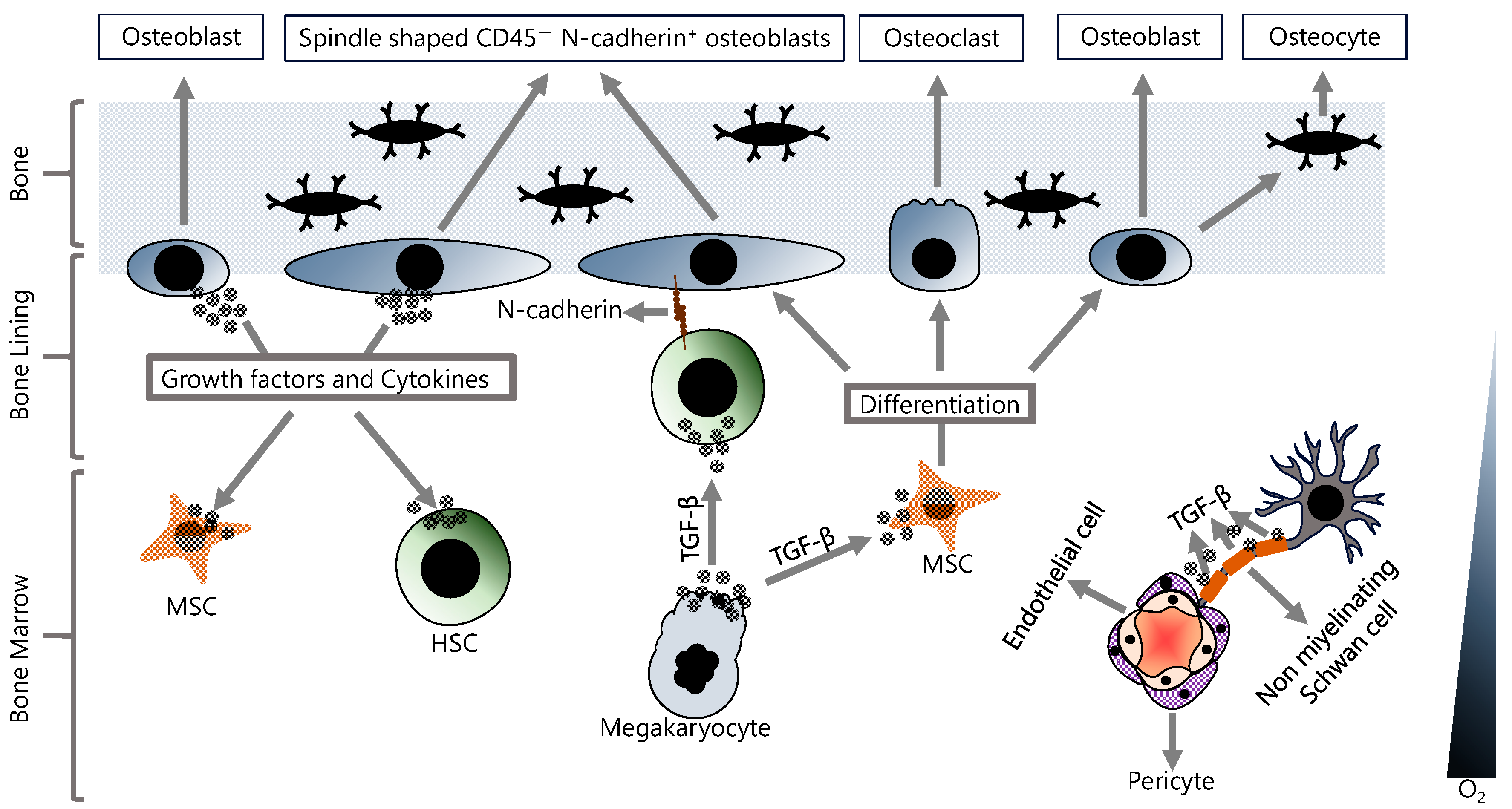
Figure 1. Hematopoietic and non-hematopoietic cells in the BM. In the BM, stem cell properties and differentiation signals are regulated by various cellular and non-cellular components, and their fine-tuned interactions are critical for maintaining homeostasis.
At least two HSC niches have been identified in BM, an endosteal niche (also known as osteogenic niche) and a perivascular niche (also known as central niche) [40]. The vascular niche has a very rich vascularization consisting of arterioles, and the cells directly associated with the vasculature can be summarized as endothelial cells, perivascular stromal cells, peri-arteriolar Ng2+ cells, non-myelinating Schwann cells, megakaryocytes, and HSCs [41]. The endosteal niche has a hypoxic environment and this low-oxygen milieu allows HSCs to remain in a quiescent state, thus maintaining the stem cell pool [42]. The endosteal niche is physically close to the trabecular bone and contains many mesenchymal stem cells (MSCs) and cells of the osteoblastic lineage, including osteoblasts, osteocytes, and osteoclasts [43]. The MSCs are the multipotent stem cells with the ability to both self-renew and differentiate. Osteoblast cells differentiate from MSCs and play an important role in bone development [44]. Osteoblasts in the endosteal niche have been shown to express many cytokines and growth factors to support the stem cell population in the BM [45]. In fact, the osteoblast cell population in the endosteal niche is not uniform, and there are at least two subpopulations of osteoblasts; (1) osteoblasts that function in bone formation and (2) spindle-shaped CD45− osteoblasts that express N-cadherin [46][47]. Overall, osteoblasts play a critical role in establishing and maintaining an essential niche microenvironment and are also involved in regulating stem cell quiescence and proliferation, in addition to bone formation.
HSCs in the BM niche are under both endogenous and exogenous stress conditions, and these stress factors can lead to DNA damage that may result in mutations if not properly repaired, and consequently, the accumulation of mutations may result in malignant transformation [48]. The malignantly transformed HSCs are referred to as leukemic stem cells (LSCs), and the gene expression profile of these cells, in addition to their mutational status, is very different from that of HSCs [49]. In addition, it has been reported that leukemia cells also secrete TGF-β, and autocrine TGF-β signaling may lead to phenotypic variation, which may be the main cause of leukemia cell heterogeneity [50]. Although LSCs have self-renewal and differentiation abilities like HSCs, these cells are abnormal and have the capacity to initiate leukemia; therefore, they are also called leukemia-initiating cells (LICs) [51][52]. Leukemias are a heterogeneous class of hematologic malignancies that result from the proliferation of immature and non-functional leukocytes, called blasts, in the BM and are classified as acute or chronic leukemias depending on the proportions of these abnormal leukocytes in the BM [53][54]. The malignant transformation of HSCs into LSCs changes the BM niche environment into a new milieu that supports leukemogenesis [55]. These changes are at least partially associated with signals from leukemic cells, and these events consequently further support leukemic cells [56][57]. In fact, changes in the structure and nature of the niche contribute to the creation of an environment that does not support normal hematopoiesis but contributes to disease progression [58]. The altered niche environment induces further damage to HSCs, transforming them into pre-leukemic and leukemic cells. Furthermore, it has been shown that the dysfunction of osteolineage cells in the BM niche can induce myelodysplasia and leukemia [57]. Consequently, the LSC-induced microenvironment creates novel environmental conditions that protect leukemia cells from chemotherapy [59]. For example, classical chemotherapeutic agents that cause DNA damage or spindle poisoning target actively proliferating cells and therefore cannot be used effectively against quiescent LSCs in the BM niche [60]. The interactions between LSCs and BM niches are important for leukemic cells, and these associations may control many vital mechanisms, including the promotion of survival, inhibition of apoptosis, and resistance to chemotherapy [61].
4. E-Cadherin and N-Cadherin, the Main EMT Markers, Have Crucial Roles in BM Homeostasis and Hematopoiesis, and Also in Leukemogenesis
EMT is an important cellular process characterized by the impairment of cell–cell adhesion properties and is associated with poor prognosis in cancer [62]. It is generally characterized by a decrease in E-cadherin expression and an increase in N-cadherin expression [63]. Although cadherin molecules are normally involved in cell–cell connections, they also have important roles in the interaction between cancer cells and the tumor microenvironment in solid tumors. In the context of hematologic malignancies, cadherin proteins are important, like solid tumors, and play a role in the interaction between leukemic cells and BM stromal cells.
5. β-Catenin in Acute Leukemia
β-catenin plays an important role in both the establishment and stability of adherens junctions by binding to cadherin proteins, thus contributing to cell–cell junctions, and increased β-catenin activity has been shown to induce EMT [64]. β-catenin is a multifunctional protein localized to the nucleus, cytosol, and centrosomes in addition to adherens junctions [65], and its levels are tightly regulated mainly by GSK3β-dependent phosphorylation and subsequent destruction by proteasomal degradation mechanisms [66]. In fact, the cytosolic β-catenin level is normally low in cells because excess β-catenin is rapidly targeted by the proteasome in a ubiquitin-dependent manner [67]. However, inhibition of GSK3β leads to accumulation of β-catenin in cells and, thus, to its activation [66]. In addition, the cellular level and the activity of β-catenin may also be regulated by Wnt-independent mechanisms including PI3K/AKT [68]. β-catenin activation can be defined as the interaction of β-catenin with co-regulators in the nucleus, which is followed by its binding to the promoters of target genes together with the co-regulators, and consequently, the controlling of the transcription of these genes.
In this context, nuclear β-catenin has been shown to interact with CREB-binding protein/E1A-binding protein p300 (CBP/p300) transcriptional co-activators and other basal transcriptional machinery apparatus member proteins [69]. In addition, β-catenin can bind to the nuclear TFs TCF/LEF and mediate the transcription of genes involved in cell proliferation such as ccnd1 (encodes cyclin D1) and Myc [70]. Aberrant β-catenin activity can result from a variety of mechanisms, including epigenetic alterations, defects in upstream activating signals such as Wnt and AKT, activating mutations in β-catenin, mutations in the GSK3β/APC/Axin complex, increased hematological and neurological expressed 1 (HN1) level and the interaction status of β-catenin with cadherins [71][72][73][74] (Figure 2). In this context, although the status of E-cadherin and N-cadherin is generally considered in terms of the migratory abilities of epithelial cells by relating to adherens junctions, they also interact with β-catenin in the cytoplasm, and this interaction contributes to the regulation of both the cellular level and intracellular localization of β-catenin [75]. In addition, various cell-specific defects can also induce abnormal β-catenin activity, such as flt3 mutations in AML. flt3 internal tandem duplication is a common defect in AML and is associated with poor prognosis [76][77]. This mutation was shown to increase the β-catenin level, thereby promoting TCF/LEF-dependent transcription [78] (Figure 2).
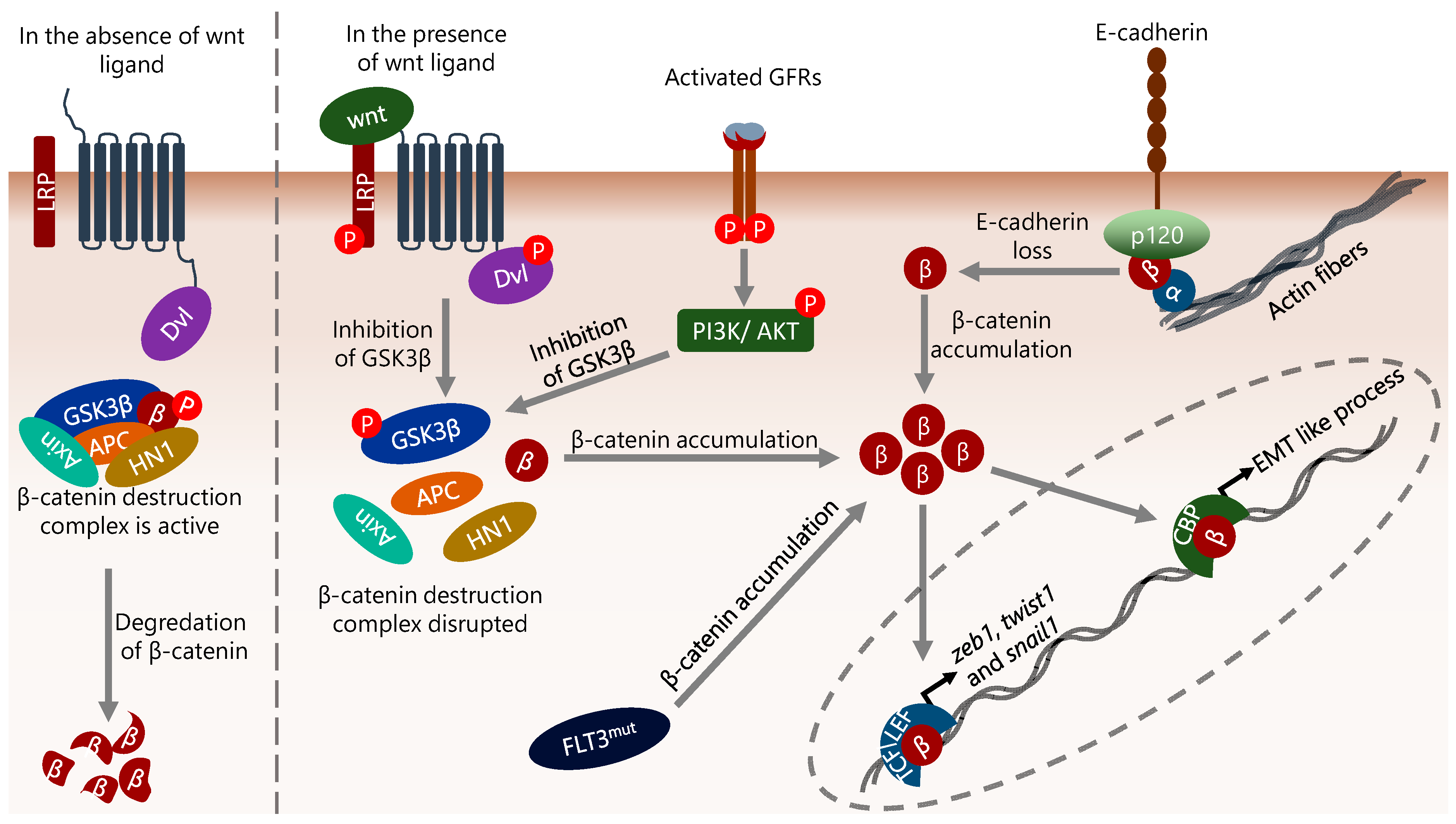
Figure 2. The diversity of mechanisms that influence β-catenin activity. β-catenin is important in the induction of EMT, and its level is mainly regulated by proteasomal degradation mechanisms in a GSK3β complex-dependent manner. Disruption of the GSK3β complex by Wnt or growth factor receptor (GFR), flt3 mutation (internal tandem duplication), or reduction/loss of E-cadherin results in cytoplasmic accumulation of β-catenin. Cytoplasmic β-catenin translocates to the nucleus, binds directly to the promoter of target genes, and consequently regulates their expression. In the context of EMT, the genes encoding EMT-inducing TFs such as zeb1, twist1 and snail1 are targets of β-catenin. In addition, β-catenin binds to the promoter of genes that induce EMT-like processes in cells and increases their transcription. α, β, and p120 represent α-catenin, β-catenin, and p120-catenin, respectively.
Aberrant activation of β-catenin in HSCs has been shown to lead to cell cycle entry and subsequent exhaustion of HSCs in BM [79]. In this context, the co-activators that interact with β-catenin are important for stem cell functions, and CBP and p300 were shown to be involved in self-renewal and differentiation of HSCs, respectively [80]. In fact, the effects of CBP and p300 in this way are not restricted to HSCs, and they act in a similar way in ESCs [81]. However, although CBP and p300 have been identified as bimodal regulators of β-catenin signaling and transcriptional activity, it is unclear to what extent β-catenin interactions with CBP or p300 affect cell fate, i.e., self-renewal or differentiation [82]. It has also been shown that β-catenin is associated with CBP in the nuclei of cells undergoing an EMT-like process [83]. However, it appears that the effect of β-catenin in inducing EMT is not limited to interaction with CBP. For example, the β-catenin/TCF4 complex has been shown to bind directly to the zeb1 promoter, increasing its expression and thereby inducing EMT [84][85]. On the other hand, it was also shown that EMT-promoting TFs can induce β-catenin expression. In this context, miR-200a, which represses zeb1/2 and snail2 expression and consequently inhibits EMT, also inhibits β-catenin expression in a ZEB1/2-dependent manner [86]. β-catenin can also bind to the promoters of genes encoding other EMT-inducing TFs, such as snail1 and twist1, in a complex with TCF/LEF family proteins, and regulate their transcription [87]. It has also been shown that β-catenin, which has a mutation that allows it to localize to the nucleus, causes a decrease in the levels of some cell–cell junction molecules, including E-cadherin, and consequently induces EMT in colon cancer cells [88]. Although both studies were performed in non-hematologic cells, their results may represent a general mechanism for β-catenin-induced EMT, including HSCs and LSCs. The effect of β-catenin on HSCs appears to be dose-dependent. In this context, it has been reported that mildly activated β-catenin can increase the clonogenicity and myeloid development of HSCs, whereas highly increased activity causes a disruption of HSC functions such as self-renewal and differentiation [89][90]. Increased β-catenin levels have been demonstrated in many cancers [69], and there are numerous reports that aberrant activation of β-catenin is involved in the pathogenesis of many hematologic malignancies, including leukemia [91]. It has been shown that β-catenin is also involved in the transformation of healthy HSCs into LSCs [92]. Although the mechanism of transformation of HSCs into LSCs is not fully understood, the critical role of β-catenin and related genes in the pathogenesis of hematological malignancies has long been observed and discussed [91].
β-catenin has been shown to be required for the development of AML and ALL LSCs [93][94], and an activating mutation in β-catenin alters the differentiation potential of myeloid progenitors and consequently causes AML development [95]. Consistently, the β-catenin level is significantly increased in AML cells, accompanied by a decrease in the E-cadherin level, and this event has been associated with poor prognosis [96][97][98][99]. Furthermore, the level of β-catenin has been shown to be higher in samples from relapsed AML patients and in BM-resident leukemic cells compared to samples from circulating blasts [100]. Accordingly, disruption of β-catenin signaling has been shown to have a potent effect against AML CSCs and also to have a synergistic effect with FLT3 inhibition on the flt3-mutant AML cells [100]. However, it has also been reported that genetic deletion of β-catenin in LSCs does not affect their ability to self-renew, contrary to previous reports [101]. To this point, it should be noted that although many independent studies have shown the strong implication of β-catenin in the transformation of HSCs, and thereby the development of LSCs, the aberrant expression or activation of β-catenin as a single factor is not sufficient for the development of leukemia [93][102][103]. On the other hand, moderate β-catenin activity in the stromal cells is also required for a balanced microenvironment that supports healthy hematopoiesis in the BM. In this context, β-catenin depletion in BM stromal cells has been shown to cause a decrease in HSC maintenance, whereas increased β-catenin results in an enhancement of HSC functions such as self-renewal [104][105]. Similarly, constitutively active β-catenin in osteoblasts residing in the BM niche impairs hematopoiesis and drives leukemogenesis [95]. Consistent with this, it has been shown that more than 35% of AML patients have increased β-catenin activity in osteoblasts in the BM [95].
References
- Fedele, M.; Sgarra, R.; Battista, S.; Cerchia, L.; Manfioletti, G. The Epithelial-Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. Int. J. Mol. Sci. 2022, 23, 800.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890.
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8.
- Imodoye, S.O.; Adedokun, K.A.; Muhammed, A.O.; Bello, I.O.; Muhibi, M.A.; Oduola, T.; Oyenike, M.A. Understanding the Complex Milieu of Epithelial-Mesenchymal Transition in Cancer Metastasis: New Insight Into the Roles of Transcription Factors. Front. Oncol. 2021, 11, 762817.
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352.
- Xia, Y.; Inoue, K.; Du, Y.; Baker, S.J.; Reddy, E.P.; Greenblatt, M.B.; Zhao, B. TGFbeta reprograms TNF stimulation of macrophages towards a non-canonical pathway driving inflammatory osteoclastogenesis. Nat. Commun. 2022, 13, 3920.
- Lambropoulou, M.; Tamiolakis, D.; Venizelos, I.; Alexiadis, G.; Anastasopoulos, G.; Limberis, V.; Galazios, G.; Tsikouras, P.; Simopoulou, M.; Nikolaidou, S.; et al. Induction of hepatic haematopoiesis with fibronectin expression by EMT stromal cells during the second trimester of development. Clin. Exp. Med. 2007, 7, 115–121.
- Goossens, S.; Janzen, V.; Bartunkova, S.; Yokomizo, T.; Drogat, B.; Crisan, M.; Haigh, K.; Seuntjens, E.; Umans, L.; Riedt, T.; et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood 2011, 117, 5620–5630.
- Wang, J.; Farkas, C.; Benyoucef, A.; Carmichael, C.; Haigh, K.; Wong, N.; Huylebroeck, D.; Stemmler, M.P.; Brabletz, S.; Brabletz, T.; et al. Interplay between the EMT transcription factors ZEB1 and ZEB2 regulates hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity. PLoS Biol. 2021, 19, e3001394.
- Li, J.; Riedt, T.; Goossens, S.; Carrillo Garcia, C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gutgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129, 460–472.
- Palen, K.; Weber, J.; Dwinell, M.B.; Johnson, B.D.; Ramchandran, R.; Gershan, J.A. E-cadherin re-expression shows in vivo evidence for mesenchymal to epithelial transition in clonal metastatic breast tumor cells. Oncotarget 2016, 7, 43363–43375.
- Lima, C.R.; Gomes, C.C.; Santos, M.F. Role of microRNAs in endocrine cancer metastasis. Mol. Cell. Endocrinol. 2017, 456, 62–75.
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184.
- Zhao, C.; Mo, L.; Li, C.; Han, S.; Zhao, W.; Liu, L. FOXN3 suppresses the growth and invasion of papillary thyroid cancer through the inactivation of Wnt/beta-catenin pathway. Mol. Cell. Endocrinol. 2020, 515, 110925.
- Carriere, P.; Calvo, N.; Novoa Diaz, M.B.; Lopez-Moncada, F.; Herrera, A.; Torres, M.J.; Alonso, E.; Gandini, N.A.; Gigola, G.; Contreras, H.R.; et al. Role of SPARC in the epithelial-mesenchymal transition induced by PTHrP in human colon cancer cells. Mol. Cell. Endocrinol. 2021, 530, 111253.
- Varisli, L.; Tolan, V. Increased ROS alters E-/N-cadherin levels and promotes migration in prostate cancer cells. Bratisl. Lek. Listy 2022, 123, 752–757.
- Yoshida, G.J.; Saya, H. Molecular pathology underlying the robustness of cancer stem cells. Regen. Ther. 2021, 17, 38–50.
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518.
- Moreb, J.S. Aldehyde dehydrogenase as a marker for stem cells. Curr. Stem Cell Res. Ther. 2008, 3, 237–246.
- Yasuda, T.; Ishimoto, T.; Baba, H. Conflicting metabolic alterations in cancer stem cells and regulation by the stromal niche. Regen. Ther. 2021, 17, 8–12.
- Kamble, D.; Mahajan, M.; Dhat, R.; Sitasawad, S. Keap1-Nrf2 Pathway Regulates ALDH and Contributes to Radioresistance in Breast Cancer Stem Cells. Cells 2021, 10, 83.
- Mori, Y.; Yamawaki, K.; Ishiguro, T.; Yoshihara, K.; Ueda, H.; Sato, A.; Ohata, H.; Yoshida, Y.; Minamino, T.; Okamoto, K.; et al. ALDH-Dependent Glycolytic Activation Mediates Stemness and Paclitaxel Resistance in Patient-Derived Spheroid Models of Uterine Endometrial Cancer. Stem Cell Rep. 2019, 13, 730–746.
- Dancik, G.M.; Varisli, L.; Tolan, V.; Vlahopoulos, S. Aldehyde Dehydrogenase Genes as Prospective Actionable Targets in Acute Myeloid Leukemia. Genes 2023, 14, 1807.
- Matsumoto, T.; Yokoi, A.; Hashimura, M.; Oguri, Y.; Akiya, M.; Saegusa, M. TGF-beta-mediated LEFTY/Akt/GSK-3beta/Snail axis modulates epithelial-mesenchymal transition and cancer stem cell properties in ovarian clear cell carcinomas. Mol. Carcinog. 2018, 57, 957–967.
- Man, Y.; Yao, X.; Yang, T.; Wang, Y. Hematopoietic Stem Cell Niche During Homeostasis, Malignancy, and Bone Marrow Transplantation. Front. Cell Dev. Biol. 2021, 9, 621214.
- Lucas, D. Structural organization of the bone marrow and its role in hematopoiesis. Curr. Opin. Hematol. 2021, 28, 36–42.
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320.
- Hinge, A.; Filippi, M.D. Deconstructing the Complexity of TGFbeta Signaling in Hematopoietic Stem Cells: Quiescence and Beyond. Curr. Stem Cell Rep. 2016, 2, 388–397.
- Yamazaki, S.; Ema, H.; Karlsson, G.; Yamaguchi, T.; Miyoshi, H.; Shioda, S.; Taketo, M.M.; Karlsson, S.; Iwama, A.; Nakauchi, H. Nonmyelinating Schwann Cells Maintain Hematopoietic Stem Cell Hibernation in the Bone Marrow Niche. Cell 2011, 147, 1146–1158.
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326.
- Blank, U.; Karlsson, S. TGF-β signaling in the control of hematopoietic stem cells. Blood 2015, 125, 3542–3550.
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320.
- Tabe, Y.; Shi, Y.X.; Zeng, Z.; Jin, L.; Shikami, M.; Hatanaka, Y.; Miida, T.; Hsu, F.J.; Andreeff, M.; Konopleva, M. TGF-beta-Neutralizing Antibody 1D11 Enhances Cytarabine-Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS ONE 2013, 8, e62785.
- Yang, Z.; Sui, L.; Toh, W.S.; Lee, E.H.; Cao, T. Stage-dependent effect of TGF-beta1 on chondrogenic differentiation of human embryonic stem cells. Stem Cells Dev. 2009, 18, 929–940.
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236.
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841.
- Fröbel, J.; Landspersky, T.; Percin, G.; Schreck, C.; Rahmig, S.; Ori, A.; Nowak, D.; Essers, M.; Waskow, C.; Oostendorp, R.A.J. The Hematopoietic Bone Marrow Niche Ecosystem. Front. Cell Dev. Biol. 2021, 9, 705410.
- Ashok, D.; Polcik, L.; Dannewitz Prosseda, S.; Hartmann, T.N. Insights Into Bone Marrow Niche Stability: An Adhesion and Metabolism Route. Front. Cell Dev. Biol. 2021, 9, 798604.
- Grenier, J.M.P.; Testut, C.; Fauriat, C.; Mancini, S.J.C.; Aurrand-Lions, M. Adhesion Molecules Involved in Stem Cell Niche Retention During Normal Haematopoiesis and in Acute Myeloid Leukaemia. Front. Immunol. 2021, 12, 756231.
- Acar, M.; Kocherlakota, K.S.; Murphy, M.M.; Peyer, J.G.; Oguro, H.; Inra, C.N.; Jaiyeola, C.; Zhao, Z.; Luby-Phelps, K.; Morrison, S.J. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 2015, 526, 126–130.
- May, M.; Slaughter, A.; Lucas, D. Dynamic Regulation of Hematopoietic Stem Cells by Bone Marrow Niches. Curr. Stem Cell Rep. 2018, 4, 201–208.
- Bruno, S.; Mancini, M.; De Santis, S.; Monaldi, C.; Cavo, M.; Soverini, S. The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 6857.
- Le, P.M.; Andreeff, M.; Battula, V.L. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018, 103, 1945–1955.
- Sugiyama, T.; Nagasawa, T. Bone Marrow Niches for Hematopoietic Stem Cells and Immune Cells. Inflamm. Allergy-Drug Targets 2012, 11, 201–206.
- Galán-Díez, M.; Kousteni, S. The Osteoblastic Niche in Hematopoiesis and Hematological Myeloid Malignancies. Curr. Mol. Biol. Rep. 2017, 3, 53–62.
- Hauge, E.M.; Qvesel, D.; Eriksen, E.F.; Mosekilde, L.; Melsen, F. Cancellous Bone Remodeling Occurs in Specialized Compartments Lined by Cells Expressing Osteoblastic Markers. J. Bone Miner. Res. 2009, 16, 1575–1582.
- Sugiyama, T.; Omatsu, Y.; Nagasawa, T. Niches for hematopoietic stem cells and immune cell progenitors. Int. Immunol. 2019, 31, 5–11.
- Bakker, S.T.; Passegué, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923.
- Velten, L.; Story, B.A.; Hernández-Malmierca, P.; Raffel, S.; Leonce, D.R.; Milbank, J.; Paulsen, M.; Demir, A.; Szu-Tu, C.; Frömel, R.; et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat. Commun. 2021, 12, 1366.
- Shingai, Y.; Yokota, T.; Okuzaki, D.; Sudo, T.; Ishibashi, T.; Doi, Y.; Ueda, T.; Ozawa, T.; Nakai, R.; Tanimura, A.; et al. Autonomous TGFbeta signaling induces phenotypic variation in human acute myeloid leukemia. Stem Cells 2021, 39, 723–736.
- Schepers, K.; Campbell, T.B.; Passegue, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267.
- Dick, J.E. Acute Myeloid Leukemia Stem Cells. Ann. N. Y. Acad. Sci. 2009, 1044, 1–5.
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228.
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719.
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol. 2017, 106, 45–54.
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013, 13, 285–299.
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857.
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff , M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298.
- Parker, J.; Hockney, S.; Blaschuk, O.W.; Pal, D. Targeting N-cadherin (CDH2) and the malignant bone marrow microenvironment in acute leukaemia. Expert Rev. Mol. Med. 2023, 25, e16.
- Ebinger, S.; Ozdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862.
- Barwe, S.P.; Quagliano, A.; Gopalakrishnapillai, A. Eviction from the sanctuary: Development of targeted therapy against cell adhesion molecules in acute lymphoblastic leukemia. Semin. Oncol. 2017, 44, 101–112.
- Greaves, D.; Calle, Y. Epithelial Mesenchymal Transition (EMT) and Associated Invasive Adhesions in Solid and Haematological Tumours. Cells 2022, 11, 649.
- Chou, Y.S.; Yang, M.H. Epithelial-mesenchymal transition-related factors in solid tumor and hematological malignancy. J. Chin. Med. Assoc. 2015, 78, 438–445.
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634.
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736.
- Wu, D.; Pan, W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010, 35, 161–168.
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898.
- Aktary, Z.; Bertrand, J.U.; Larue, L. The WNT-less wonder: WNT-independent beta-catenin signaling. Pigment. Cell Melanoma Res. 2016, 29, 524–540.
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/beta-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307.
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165.
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3.
- Koni, M.; Pinnaro, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697.
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196.
- Varisli, L.; Ozturk, B.E.; Akyuz, G.K.; Korkmaz, K.S. HN1 negatively influences the beta-catenin/E-cadherin interaction, and contributes to migration in prostate cells. J. Cell Biochem. 2015, 116, 170–178.
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305.
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322.
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880.
- Tickenbrock, L.; Schwable, J.; Wiedehage, M.; Steffen, B.; Sargin, B.; Choudhary, C.; Brandts, C.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005, 105, 3699–3706.
- Kirstetter, P.; Anderson, K.; Porse, B.T.; Jacobsen, S.E.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056.
- Zhu, Y.; Wang, Z.; Li, Y.; Peng, H.; Liu, J.; Zhang, J.; Xiao, X. The Role of CREBBP/EP300 and Its Therapeutic Implications in Hematological Malignancies. Cancers 2023, 15, 1219.
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673.
- Li, J.; Sutter, C.; Parker, D.S.; Blauwkamp, T.; Fang, M.; Cadigan, K.M. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 2007, 26, 2284–2294.
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314.
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209.
- Sanchez-Tillo, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Gyorffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257.
- Su, J.; Zhang, A.; Shi, Z.; Ma, F.; Pu, P.; Wang, T.; Zhang, J.; Kang, C.; Zhang, Q. MicroRNA-200a suppresses the Wnt/beta-catenin signaling pathway by interacting with beta-catenin. Int. J. Oncol. 2012, 40, 1162–1170.
- Howe, L.R.; Watanabe, O.; Leonard, J.; Brown, A.M. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003, 63, 1906–1913.
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.B.; Jung, S.H.; Choi, H.J.; et al. beta-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019, 9, 18440.
- Luis, T.C.; Naber, B.A.; Roozen, P.P.; Brugman, M.H.; de Haas, E.F.; Ghazvini, M.; Fibbe, W.E.; van Dongen, J.J.; Fodde, R.; Staal, F.J. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 2011, 9, 345–356.
- Famili, F.; Brugman, M.H.; Taskesen, E.; Naber, B.E.A.; Fodde, R.; Staal, F.J.T. High Levels of Canonical Wnt Signaling Lead to Loss of Stemness and Increased Differentiation in Hematopoietic Stem Cells. Stem Cell Rep. 2016, 6, 652–659.
- Chiarini, F.; Paganelli, F.; Martelli, A.M.; Evangelisti, C. The Role Played by Wnt/beta-Catenin Signaling Pathway in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1098.
- Lento, W.; Congdon, K.; Voermans, C.; Kritzik, M.; Reya, T. Wnt Signaling in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a008011.
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/β-Catenin Pathway Is Required for the Development of Leukemia Stem Cells in AML. Science 2010, 327, 1650–1653.
- Gekas, C.; D’Altri, T.; Aligue, R.; Gonzalez, J.; Espinosa, L.; Bigas, A. beta-Catenin is required for T-cell leukemia initiation and MYC transcription downstream of Notch1. Leukemia 2016, 30, 2002–2010.
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244.
- Bao, B.X.; An, X.Z.; Li, P.F.; Li, Y.J.; Cui, Y.H.; Tang, X.; Liu, Q.H.; Hu, Y.N.; Liu, W.; Liang, S.Y.; et al. . Zhongguo Shi Yan Xue Ye Xue Za Zhi 2019, 27, 339–347.
- Wagstaff, M.; Coke, B.; Hodgkiss, G.R.; Morgan, R.G. Targeting beta-catenin in acute myeloid leukaemia: Past, present, and future perspectives. Biosci. Rep. 2022, 42, BSR20211841.
- Yu, S.; Han, R.; Gan, R. The Wnt/beta-catenin signalling pathway in Haematological Neoplasms. Biomark. Res. 2022, 10, 74.
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403.
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/beta-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2417–2429.
- Zhao, X.; Shao, P.; Gai, K.; Li, F.; Shan, Q.; Xue, H.H. beta-catenin and gamma-catenin are dispensable for T lymphocytes and AML leukemic stem cells. eLife 2020, 9, e55360.
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618.
- Perry, J.M.; Tao, F.; Roy, A.; Lin, T.; He, X.C.; Chen, S.; Lu, X.; Nemechek, J.; Ruan, L.; Yu, X.; et al. Overcoming Wnt-beta-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol. 2020, 22, 689–700.
- Nemeth, M.J.; Mak, K.K.; Yang, Y.; Bodine, D.M. beta-Catenin expression in the bone marrow microenvironment is required for long-term maintenance of primitive hematopoietic cells. Stem Cells 2009, 27, 1109–1119.
- Kim, J.A.; Kang, Y.J.; Park, G.; Kim, M.; Park, Y.O.; Kim, H.; Leem, S.H.; Chu, I.S.; Lee, J.S.; Jho, E.H.; et al. Identification of a stroma-mediated Wnt/beta-catenin signal promoting self-renewal of hematopoietic stem cells in the stem cell niche. Stem Cells 2009, 27, 1318–1329.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
470
Revisions:
2 times
(View History)
Update Date:
20 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No