 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robert Melchers | -- | 4952 | 2024-02-09 04:49:57 | | | |
| 2 | Lindsay Dong | Meta information modification | 4952 | 2024-02-11 12:40:26 | | |
Video Upload Options
Corrosion on the interface between a metal alloy, such as steel, and a wet, permeable non-metallic medium is of considerable practical interest. Examples include the interface between steel and water, the atmosphere or concrete, as for steel reinforcement bars; between metal and soil, as for buried cast iron or steel pipes; deposits of some type, as in under-deposit corrosion; and the interface with insulation, protective coatings, or macro- or micro-biological agents. In all cases, corrosion initiation depends on the characteristics of the interfacial zone, both of the metal and the medium, and the spatial variability. For (near-)homogeneous semi-infinite media with good interfacial contact, the pitting, crevices and general corrosion of the metal will be largely controlled by the metal (micro-)characteristics, including its inclusions, imperfections and surface roughness.
1. Introduction
2. Classical Models for Corrosion Initiation and Early Development
2.1. ‘Pure’ Metal in Contact with a Wet Homogeneous (Pure) Medium
2.2. Practical Metals and a Wet Homogeneous (Pure) Medium
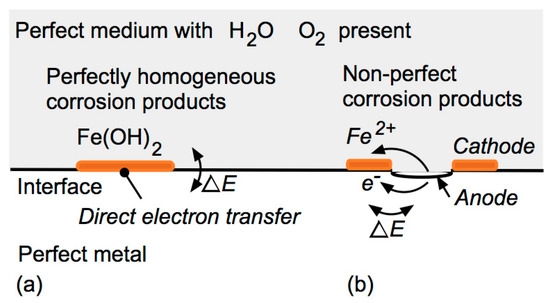
2.3. Effect of Chlorides in a Wet Homogeneous Medium
3. Corrosion Initiation for Non-Homogeneous Media
3.1. Overview
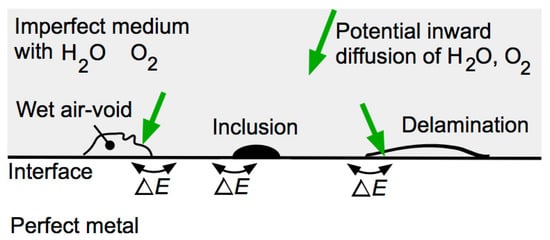
3.2. Corrosion Initiation for Ferrous Metals in Contact with Soils
3.3. Corrosion Initiation for Reinforcing Steel in Concrete
3.4. Corrosion Initiation under Protective Coatings
4. Development of Corrosion and Pitting with Continued Exposure
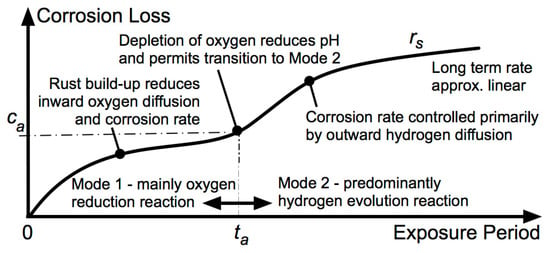
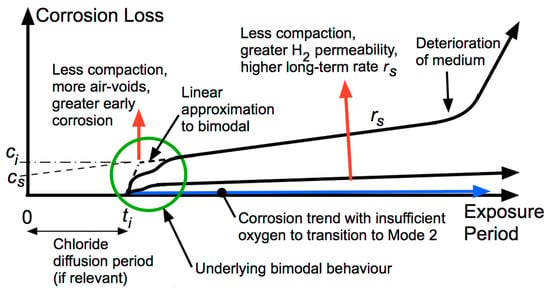
5. Long-Term Corrosion
6. Conclusions
-
Both local differences in electrochemical potential due to inhomogeneities of the steel and inhomogeneities of the surface of the medium interfacing with the steel can be sources of initiation of corrosion for the steel, although, for many practical applications, the inhomogeneities of the medium are of greater importance due to their greater physical scale.
-
Corrosion of steel in water and the atmosphere may be considered specialized cases of corrosion in homogeneous media extending over semi-infinite spaces.
-
As corrosion at the interface progresses, the instantaneous corrosion rate is increasingly governed by the properties of the corrosion products at the interface and less by the properties of the steel or the medium, with corrosion progressing eventually to and then through the hydrogen evolution as the principal long-term cathodic reaction in all cases.
-
The same mechanisms are likely to hold for other media of types similar to those considered herein when in contact with alloys and metals other than steel and for which the hydrogen evolution reaction is feasible.
-
Breakdown of the medium itself may lead to steel corrosion—it is not always the case that the corrosion of the steel causes damage to, and possibly breakdown of, the medium.
References
- Ackland, B.G.; Dylejko, K.P. Critical questions and answers about cathodic protection. Corros. Eng. Sci. Technol. 2019, 54, 688–697.
- Coogan, C. Marine Corrosion and Cathodic Protection; CRC Press: Boca Raton, FL, USA, 2022.
- Jones, D.A. Principles and Prevention of Corrosion, 2nd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1996.
- Garrels, R.M.; Christ, C.L. Solutions, Minerals and Equilibria; Harper & Row: New York, NY, USA, 1965.
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions; NACE Int.: Houston, TX, USA, 1974.
- Mercer, A.D.; Lumbard, E.A. Corrosion of mild steel in water. Br. Corros. J. 1995, 30, 43–55.
- Abiko, K.; Nakajima, T.; Harima, N.; Takaki, S. Preparation of 10 kg ingot of ultra-pure iron. Phys. Status Solidi 1998, 167, 347–355.
- Abiko, K. Why do we study ultra-pure base metals? Mater. Trans. JIM 2000, 41, 233–237.
- Khan, L.; Sato, K.; Okuyama, S.; Kobayashi, T.; Ohashi, K.; Hirasaka, K.; Nikawa, T.; Takada, K.; Higashitani, A.; Abiko, K. Ultra-high purity iron is a novel and very compatible biomaterial. J. Mech. Behav. Biomed. Mater. 2020, 106, 103744.
- Ferrara, E.; Olivetti, E.; Fiorillo, F.; Forton, R.; Martino, L.; Rocchino, L. Microstructure and magnetic properties of pure iron for cyclotron electromagnets. J. Alloys Compd. 2014, 615, S291–S295.
- Li, S.; Hihara, L.H. The comparison of the corrosion of ultrapure iron and low-carbon steel under NaCl-electrolyte droplets. Corros. Sci. 2016, 108, 200–204.
- van der Voort, G.F. Metallurgy: Principles and Practice; McGraw-Hill: New York, NY, USA, 1984.
- Wranglen, G. Review article on the influence of sulphide inclusions on the corrodibility of Fe and steel. Corros. Sci. 1969, 9, 585–602.
- Evans, U.R. The Corrosion and Oxidation of Metals: Scientific Principles and Practical Applications; Edward Arnold: London, UK, 1960.
- Melchers, R.E.; Jeffrey, R. Surface roughness effect on marine immersion corrosion of mild steel. Corrosion 2004, 60, 697–703.
- Burstein, G.T.; Pistorius, P.C.; Mattin, S.P. The nucleation and growth of corrosion pits on stainless steels. Corros. Sci. 1993, 35, 57–62.
- Foley, R.T. Role of the chloride ion in iron corrosion. Corrosion 1970, 26, 58–70.
- Foley, R.T. Complex ions and corrosion. J. Electrochem. Soc. 1975, 11, 1493–1994.
- Heyn, E.; Bauer, O. Ueber den Angriff des Eisens durch Wasser und wässerige Losungen. Stahl. Eisen 1908, 28, 1564–1573.
- Brasher, D. Stability of the oxide film on metals in relation to inhibition of corrosion. I. Mild steel in the presence of aggressive ions. Brit. Corr. J. 1967, 2, 95–103.
- Baylis, J.R. Factors other than dissolved oxygen influencing the corrosion of iron pipes. Ind. Engrg. Chem. 1926, 18, 370–380.
- Little, B.J.; Gerke, T.L.; Lee, J.S. Mini-review: The morphology, mineralogy and microbiology of accumulated iron corrosion products. Biofouling 2014, 30, 941–948.
- Greene, N.D.; Fontana, M.G. A critical analysis of pitting corrosion. Corrosion 1959, 15, 41–47.
- Sharland, S.M.; Tasker, P.W. A mathematical model of crevice and pitting corrosion—I. The physical model. Corros. Sci. 1988, 28, 603–620.
- Wranglen, G. Pitting and sulphide inclusions in steel. Corros. Sci. 1974, 14, 331–349.
- Butler, G.; Stretton, P.; Beynon, J.G. Initiation and growth of pits on high-purity iron and its alloys with chromium and copper in neutral chloride solutions. Br. Corros. J. 1972, 7, 168–173.
- Pourbaix, M. Significance of protection potential in pitting and intergranular corrosion. Corrosion 1970, 26, 31–438.
- Romanoff, M. Underground Corrosion, National Bureau of Standards Circular 579; US Government Printing Office: Washington, DC, USA, 1957.
- Rajani, B.; Kleiner, Y. Comprehensive review of structural deterioration of water mains: Physically based models. Urban. Water 2001, 3, 151–164.
- Cole, I.S.; Marney, D. The science of pipe corrosion: A review of the literature on the corrosion of ferrous metals in soils. Corros. Sci. 2012, 56, 5–16.
- Kleiner, Y.; Rajani, B.; Krys, D. Performance of ductile iron pipes. I: Characterization of exterior corrosion patterns. J. Infrastruct. Syst. ASCE 2013, 19, 108–119.
- von Wolzogen Kuhr, C.A.H.; van der Vlugt, L.S. Graphitization of cast iron as an electrochemical process in anaerobic soils, Water (den Haag), 18: 147–165 (in Dutch). Transl. Corros. 1971 1934, 17, 293–299.
- Ricker, R.E. Analysis of pipeline steel corrosion data from NBS (NIST) studies conducted between 1922–1940 and relevance to pipeline management. J. Res. Nat. Inst. Stand. Technol. 2011, 115, 373–392.
- Melchers, R.E. Models for prediction of long-term corrosion of cast iron water mains. Corrosion 2020, 76, 441–450.
- Wichers, C.M. Korrosion asphaltierter eiserner Rohre. Das. Gas. Wasserfach 1934, 77, 131–132.
- Liengen, T.; Feron, D.; Basseguy, R.; Beech, I.B. (Eds.) Understanding Biocorrosion, European Federation of Corrosion Publications; Number 66; Woodhead Publishing in Materials: Cambridge, UK, 2014.
- Skovhus, T.L.; Eckert, R.B. (Eds.) Failure Analysis of Microbiologically Influenced Corrosion; CRC Press: Boca Raton, FL, USA, 2021.
- Richardson, M.G. Fundamentals of Durable Reinforced Concrete; Spon Press: London, UK, 2002.
- Angst, U.M.; Geiker, M.R.; Michel, A.; Gehlen, C.; Wong, H.; Isgor, O.B.; Elsener, B.; Hansson, C.M.; François, R.; Hornbostel, K.; et al. The steel-concrete interface. Mater. Struct. 2017, 50, 1–24.
- Melchers, R.E.; Chaves, I.A. A comparative study of chlorides and longer-term reinforcement corrosion. Mater. Corros. 2017, 68, 613–621.
- Wakeman, C.M.; Dockweiler, E.V.; Stover, H.E.; Whiteneck, L.L. Use of concrete in marine environments. Proc. ACI 1958, 54, 841–856.
- Melchers, R.E.; Li, C.Q. Reinforcement corrosion initiation and activation times in concrete structures exposed to severe marine environments. Cem. Concr. Res. 2009, 39, 1068–1076.
- Angst, U.M.; Elsener, B.; Larsen, C.K.; Vennesland, O. Critical chloride content in reinforced concrete—A review. Cem. Concr. Res. 2009, 39, 1122–1138.
- Berensen, A.M. Marine Painting Manual; Springer-Science & Business Media BV: Dordrecht, The Netherlands, 1989.
- Hare, C.H. Corrosion Control of Steel by Organic Coatings; Revie, R.W., Ed.; Wiley: New York, NY, USA, 2011; pp. 971–983.
- Melchers, R.E. A review of trends for corrosion loss and pit depth in longer-term exposures. Corros. Mater. Degrad. 2018, 1, 4.
- Refait, P.H.; Grolleau, A.-M.; Jeannin, M.; Rémazeilles, C.; Sabot, R. Corrosion of carbon steel in marine environments: Role of the corrosionproduct layer. Corros. Mater. Degrad. 2020, 1, 198–218.
- Melchers, R.E.; Jeffrey, R. The transition from short- to long-term marine corrosion of mild steel—1. experimental observations—2. parameterization and modeling. Corrosion 2022, 78, 415–436.
- Stratmann, M.; Bohnenkamp, K.; Engell, H.J. An electrochemical study of phase-transitions in rust layers. Corros. Sci. 1983, 23, 969–985.
- Melchers, R.E. Long-term immersion corrosion of steels in seawaters with elevated nutrient concentration. Corros. Sci. 2014, 81, 110–116.
- Melchers, R.E. Long-term durability of marine reinforced concrete structures. J. Mar. Sci. Eng. 2020, 8, 290.
- Melchers, R.E.; Chaves, I.A. Reinforcement corrosion in marine concretes—2. Long-term effects. ACI Mater. J. 2020, 117, 217–228.
- Melchers, R.E.; Howlett, C.M. Reinforcement corrosion of the Phoenix caissons after 75 years of marine exposure. Proc. Inst. Civ. Eng.-Marit. Eng. 2020, 174, 19–30.
- Melchers, R.E.; Humphrey, H. Concrete alkali-aggregate reactivity and initiation of steel reinforcement corrosion. Corros. Mater. Degrad. 2023, 4, 428–444.
- Melchers, R.E. Mechanisms in long-term marine corrosion of steel reinforcement in concrete. Corrosion 2023, 79, 380–387.
- Southwell, C.R.; Bultman, J.D.; Alexander, A.L. Corrosion of metals in Tropical environments—Final report of 16 years exposures. Mater. Perform. 1976, 15, 9–25.

