Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lijuan Chen | -- | 2822 | 2024-02-08 04:42:26 | | | |
| 2 | Catherine Yang | Meta information modification | 2822 | 2024-02-08 06:10:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Huang, E.; Wang, X.; Chen, L. Regulated Cell Death Mechanisms in Endometriosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/54890 (accessed on 25 June 2026).
Huang E, Wang X, Chen L. Regulated Cell Death Mechanisms in Endometriosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/54890. Accessed June 25, 2026.
Huang, Erqing, Xiaoli Wang, Lijuan Chen. "Regulated Cell Death Mechanisms in Endometriosis" Encyclopedia, https://encyclopedia.pub/entry/54890 (accessed June 25, 2026).
Huang, E., Wang, X., & Chen, L. (2024, February 08). Regulated Cell Death Mechanisms in Endometriosis. In Encyclopedia. https://encyclopedia.pub/entry/54890
Huang, Erqing, et al. "Regulated Cell Death Mechanisms in Endometriosis." Encyclopedia. Web. 08 February, 2024.
Copy Citation
Regulated cell death (RCD) represents a distinct mode of cell demise, differing from accidental cell death (ACD), characterized by specific signaling cascades orchestrated by diverse biomolecules. The regular process of cell death plays a crucial role in upholding internal homeostasis, acting as a safeguard against biological or chemical damage.
endometriosis
regulated cell death (RCD)
apoptosis
pyroptosis
targeted therapy
1. Apoptosis in Endometriosis
In 1972, Kerr and colleagues identified apoptosis, noting the morphological similarity of cell demise across various pathological conditions and normal tissue contexts [1]. This is the most well-characterized form of RCD, causing shrinkage, nuclear chromatin condensation, and fragmentation of the nucleus [2]. It is essential for disease prevention to maintain a balance between apoptosis and cell proliferation [3]. Cells with irreversible DNA damage are also removed by apoptosis as part of the immune system’s defense against infections [4]. By recognizing, uptaking, and degrading intact cells, apoptosis protects tissue from inflammation without releasing harmful contents. Rather than being a death mode, apoptosis is more like a mechanism for clearing out cells. Apoptosis is usually catalyzed by the proteolytic cleavage of thousands of proteins through the enzymatic activity of effector caspases like caspase 3 (Figure 1) [5]. Moreover, apoptosis can also be activated by granzyme contained within cytotoxic granules in T-cells or NK cells and through perforin-mediated pore formation in target cells [6]. In most mammalian cells, the increase in mitochondrial outer membrane permeability and the release of cytochrome c into the cytosol are key nodes that trigger apoptosis, which is regulated by pro-apoptotic and anti-apoptotic factors of the BCL-2 family [7]. Mitochondrial dysfunction can lead to impaired apoptosis. For example, in endometriotic tissues, CHCHD2 (Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 2) expression may contribute to the pathogenesis of endometriosis through its regulation of mitochondria-mediated apoptosis [8]. Some scholars have confirmed that although the expression of apoptosis-related genes varies depending on the pathological type, there is overexpression of anti-apoptotic factors and insufficient expression of pro-apoptotic factors in endometriosis [9][10]. Another extrinsic cell death pathway is achieved through pro-apoptotic receptors such as Fas, TNF, TRAIL (TNF-related apoptosis-inducing ligand), etc. [11]. In endometriosis, apoptosis seems to be a protection mechanism. Estrogen and progesterone are also involved in this process. For endometriosis, estrogen mainly inhibits apoptosis through protein kinases, NF-kB, SRC-1, and other signaling pathways. Han and his colleagues discovered that ERβ can interact with cellular apoptotic machinery in the cytoplasm to inhibit TNF-a-induced apoptosis [12]. This mechanism may help endometriosis lesions evade endogenous immune surveillance. The mechanism by which progesterone regulates endometriosis apoptosis is relatively complex. In vivo experiments, normal women supplemented with progesterone at the late secretory phase can play an anti-endometrial apoptosis role, while in vitro experiments, progesterone can induce endometrial apoptosis [13]. The specific mechanism still needs more experimental verification. Contrary to endometrial cells, which undergo reduced apoptosis, ovarian granulosa cells of endometriosis patients undergo increased apoptosis. Consequently, folliculogenesis, oocyte and embryo quality, and IVF (in vitro fertilization) outcomes could be adversely affected [14][15]. Interestingly, it was found that when follicular fluid from patients with endometriosis-associated infertility was used for granulosa cell culture in patients with simple tubal infertility, the level of granulosa cell apoptosis was significantly increased, suggesting that there is a pro-granulosa cell apoptotic phenotype in follicular fluid from patients with endometriosis [16]. Additionally, elevated apoptosis was also found in cumulus cells from patients with ovarian endometrioma [17]. The exchange of material and signals between the cumulus cell and the oocytes is necessary for the maturation and ovulation of the oocytes. It is reasonable to speculate that infertility in patients with endometriosis may be associated with increased apoptosis of the cumulus cell.
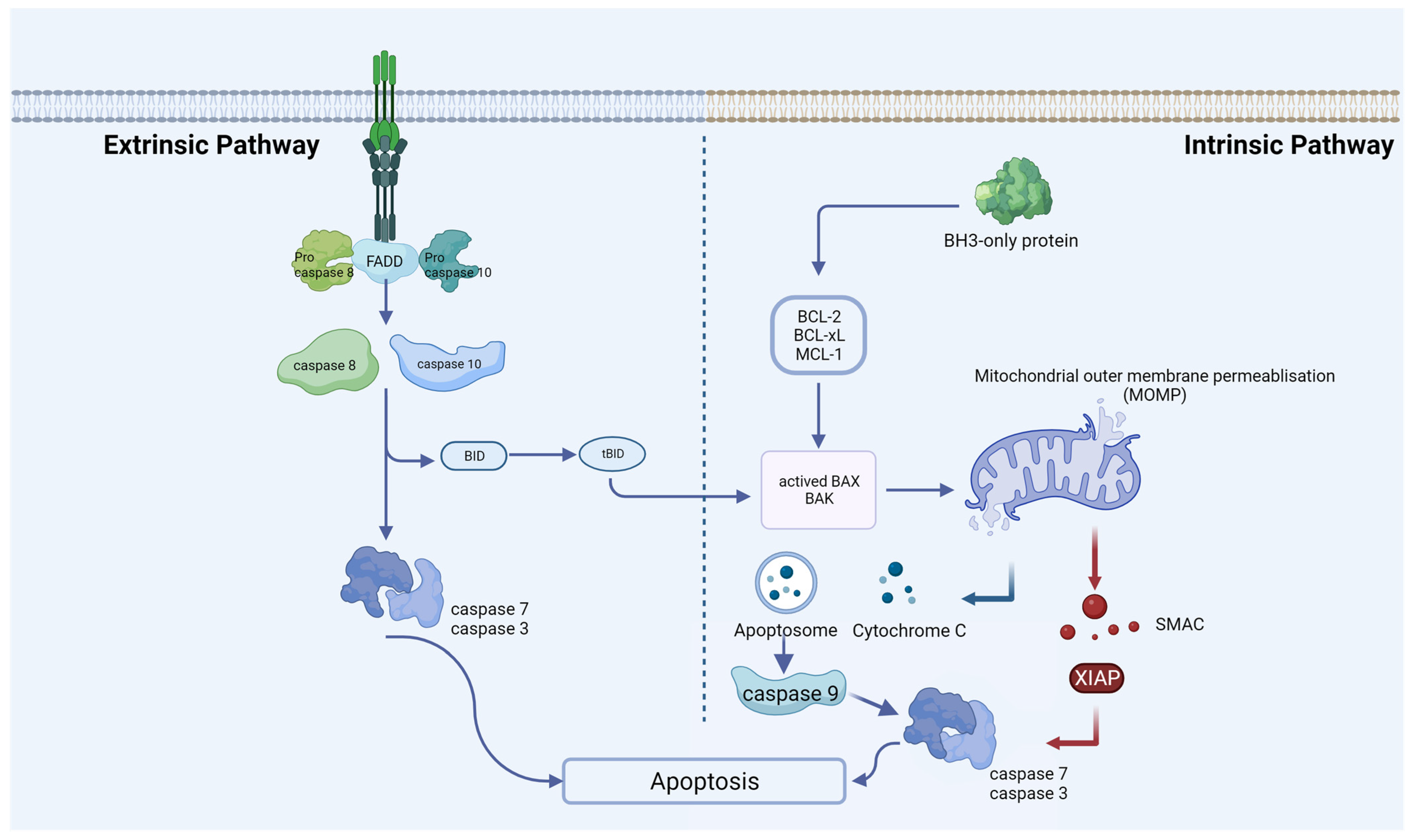
Figure 1. Extrinsic and intrinsic apoptotic pathways. An extrinsic pathway (death receptor-induced pathway) or an intrinsic pathway (mitochondrial or BCL-2-regulated pathway) can induce apoptosis. Caspases 8 and 10 undergo activation through the extrinsic pathway, a process initiated upon activation of death receptors situated at the plasma membrane. Various stress stimuli activate the intrinsic pathway, leading to mitochondrial outer-membrane permeabilization (MOMP), the release of cytochrome C, the formation of the apoptosome, and subsequent activation of caspase-9. At the proteolytic activation step of the effector caspases-3 and -7, both apoptotic pathways converge after the initiator caspases-8 and -9 are activated.
The recent research report affirms that GRIK1 antisense RNA (GRIK1-AS1) is capable of attenuating the proliferation of endometrial stromal cells. This effect is achieved through the inhibition of cell-cycle processes and the facilitation of apoptosis [18]. Another article revealed that an exosomal lncRNA, HOTAIR (HOX Transcript Antisense RNA), inhibits endometrial stromal cell apoptosis through sponging miR-761 [19]. Apoptotic activity linked with miRNA has been implicated in the pathological processes underlying endometriosis. Illustratively, a recent examination focusing on miRNAs governing adhesion and apoptosis revealed a noteworthy elevation in the expression levels of miR-93-5p and miR-7-5p in the cohorts afflicted with deep infiltrating endometriosis and endometrioma, as opposed to those presenting with lesions of superficial peritoneal endometriosis. Perhaps these results could help identify differences between pathological phenotypes of endometriosis [20].
Apoptosis is controlled by NF-κB transcription factors in a wide range of cell types, whether they block apoptosis or induce it. Endometrial cells promoted miR-138 to induce exosome-mediated inflammation and apoptosis in endometriosis through the VEGF/NF-κB signaling pathway [21]. Reactivating endometriosis apoptosis to inhibit the progression of the lesion is also a new direction for the treatment of endometriosis in the future. Ectopic lesions treated with oleuropein displayed higher levels of caspase-3 cleavage. Oleuropein also reactivated apoptosis in ectopic lesions by inhibiting ERβ and suppressing mouse endometriosis progression [22]. In addition, apoptosis is also regulated by some epigenetic modifications. For example, the depletion of histone deacetylase 2, HDAC2, can significantly promote the apoptosis of endometriosis cells [23]. Among the many studies of RCD mechanisms and endometriosis, apoptosis is undoubtedly one of the most studied and intensively researched mechanisms. Reducing or even reversing the anti-apoptotic properties of endometriosis lesions may become a completely new approach to treating endometriosis in the future.
2. Pyroptosis in Endometriosis
As a form of lytically programmed cell death, pyroptosis is initiated by inflammasomes that detect contamination or perturbation within the cytosol. Caspases-1 (canonical pathway) or caspase-11/4/5 (non-canonical pathway) are activated, which cleave gasdermin D (GSDMD) [24]. The morphological manifestation of pyroptosis is cell swelling and rupture of the plasma membrane, causing a release of pro-inflammatory cytokines and cellular contents into the extracellular space [24]. Unlike apoptosis, pyroptosis preserves mitochondrial integrity and prevents cytochrome C leakage.
Inflammasomes are protein complexes that contain three main parts: receptor proteins, adaptor proteins (ASCs), and downstream caspases. Receptor proteins are divided into the NOD-like receptor (NLR) family and the PYHIN family. Inflammasomes are assembled in response to pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs) [25]. There are four main prototypes of inflammasome sensors found to date—NLR family pyrin domain containing 1 (NLRP1), NLRC4 (NLR Family CARD Domain Containing 4), absent in melanoma-2 (AIM2), and NLRP3 (NOD-like receptor thermal protein domain associated protein 3) [26]. In the canonical pathway, when these canonical inflammasome sensors are activated, the majority of these sensors interact with the ASCs, which activate caspase 1. Caspase1 cleaves GSDMD into two fragments, one at the C-terminus and the other at the N-terminus, which causes pores in the cell membrane via lysin phosphoinositide/cardiolipin-containing liposomes and triggers pyroptosis [27]. Moreover, caspase-1 also matures pro-IL-1β and pro-IL-18 into IL-1β and IL-18, which are released through the necrotic membrane pores formed by the GSDMD N-terminal fragment [28]. Non-canonical inflammasome pathways are uniquely mediated by caspase 11 (mice) and caspase 4/5 (humans). These caspases can directly bind with LPS and conduct the cleavage of GSDMD. By contrast, non-canonical inflammasomes that activate caspase 4/5/11 proteolysis only GSDMD cannot activate IL-1β and IL-18 directly [29]. However, secondary GSDMD pore-induced membrane damage and NLRP3 activation result in cytokine maturation in addition to GSDMD processing. This process also leads to an inflammatory response.
In recent years, transcription factors have been shown to regulate pyroptosis in endometriosis. TRIM24 is a member of the three-gene sequence protein (TRIM) family and belongs to the transactivator. The TRIM24 receptor targets are located in the nucleus and affect their expression and function by regulating chromosomal remodeling-related proteins [30]. An inhibitory effect of TRIM24 was observed on the NLRP3/CASP1β-mediated pyroptosis and cell migration of human endometrial stromal cells. The upregulation of TRIM24 facilitated the ubiquitination of NLRP3 [31]. Another transcription factor, FoxA2, is expressed specifically in the glands of the uterus and is a critical regulator of postnatal uterine gland differentiation in mice [32]. It is reported that upregulation of FoxA2 (Forkhead Box A2) downregulates ERβ by transcriptionally inhibiting IGF2BP1, thereby repressing pyroptosis in endometriosis [33].
Endometriosis can induce chronic pelvic inflammation and tissue fibrosis. During pathogenesis, PGE2-induced NLRP3/caspase1 pyroptosis plays a vital role in the invasion of endometriosis lesions. Huang et al. examined the expression level of pyroptosis-related proteins such as NLRP3, caspase-1, IL-1β, and IL-18 in endometriosis and found them significantly higher than normal endometrium [34]. In a bioinformatics study on endometriosis, researchers screened for pyroptosis genes that are closely related to endometriosis and used these genes to score pyroptosis levels in samples from public databases [35]. There is a strong correlation between higher levels of pyroptosis and more aggressive disease features, including epithelial–mesenchymal transition, angiogenesis, and impaired immunity [36]. Endometriosis relies heavily on new blood and vascular system formation to progress, so angiogenesis is essential in its progression [37][38][39]. NLRP3 inflammasome-mediated activation of pyroptosis can affect angiogenesis in endometriosis in a Notch1-dependent manner [40]. Fibrosis is the development of fibrous connective tissue in response to repeated tissue injury and repair, with myofibroblasts playing a key role in driving the fibrotic process. Once myofibroblasts are activated and produce a large amount of collagen extracellular matrix, they destroy the surrounding cellular structures. Fibrotic tissue often appears as scarring that is stiff and lacks blood vessels, complicating the surgical anatomy of endometriosis. Liu’s team demonstrated that aberrantly elevated lnc-MALAT1 (Metastasis Associated Lung Adenocarcinoma Transcript 1) in ectopic endometrium is associated with NLRP3-mediated pyroptosis and fibrosis, and lnc-MALAT1 sponges miR-141-3p to promote NLRP3 expression [41]. From the above results, it can be seen that pyroptosis regulates the pathological processes of endometriosis, such as inflammatory immune response, cell invasion, and fibrosis (Figure 2). In particular, the NLRP3-mediated pyroptosis pathway is involved in many mechanisms.
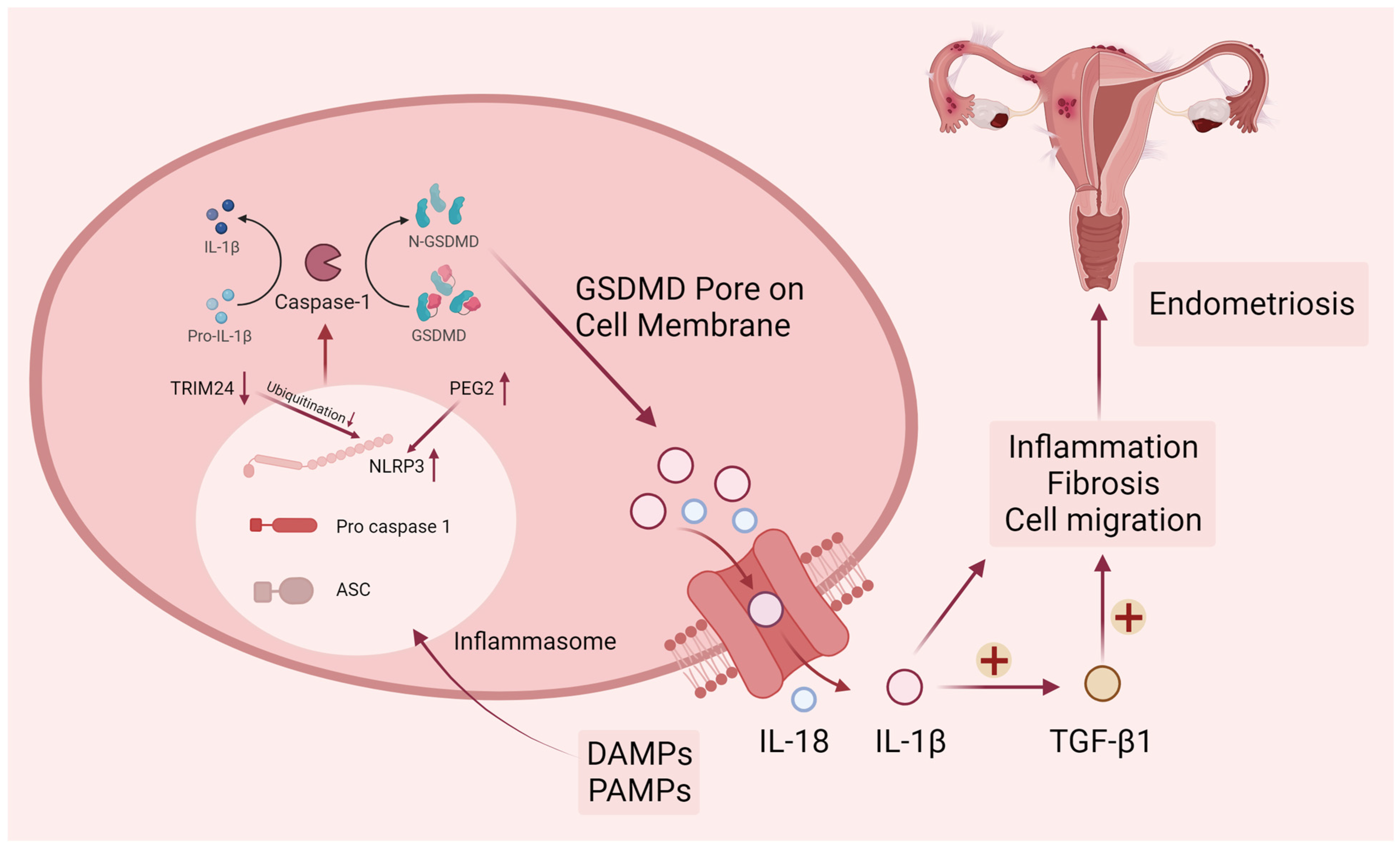
Figure 2. Canonical pyroptosis pathway in endometriosis. DAMPs (damage-associated molecular patterns) and PAMPs (pathogen-associated molecular patterns) elicit the activation of cytosolic canonical inflammasomes, notably NLRP3, leading to the subsequent cleavage of caspase-1. Following the activation of inflammatory caspases, the precursors pro-IL-1β, pro-IL-18, and GSDMD undergo cleavage. The N-terminal fragment of GSDMD (N-GSDMD) can form pores on the plasma membrane, facilitating the release of inflammatory mediators such as IL-1β and IL-18. The resultant pro-inflammatory microenvironment in endometriosis is conducive to the promotion of endometriotic cell migration and fibrosis. Upper arrows represent increased protein levels and lower arrows represent decreased protein levels.
3. Ferroptosis in Endometriosis
Iron overload and lipid peroxidation are typical symptoms of ferroptosis, a type of iron-dependent cell death [42]. Several ferroptosis-inducing factors have been identified as influencing glutathione peroxidase, which eventually leads to decreased antioxidant capacity and lipid reactive oxygen species (ROS) accumulation in cells that ultimately causes oxidative cell death [43]. Morphologically, cells undergoing ferroptosis usually show necrosis-like morphological changes. These features include a loss of plasma membrane integrity, cytoplasmic swelling, swelling of cytoplasmic organelles, and moderate chromatin condensation [44]. Ferroptosis can also be accompanied by autophagosome development and detachment.
The system Xc-GSH-GPX4 pathway is a classic pathway for ferroptosis. System Xc- is a cystine/glutamate antiporter that exchanges extracellular cystine with intracellular glutamate [45]. Once cystine enters the cell, it is rapidly reduced, producing cysteine for glutathione biosynthesis. Glutathione plays a crucial role in intracellular antioxidant defense (GSH). An increase in oxidative stress and cell death can occur when GSH is depleted [46]. GPX4 is a member of the glutathione peroxidase (GPX) family. GPX4 is the only intracellular GPX used in the reduction of liposomal peroxides, which can convert lipid hydroperoxides into non-toxic lipid alcohols and prevent ferroptosis [47]. Intracellular GSH depletion and decreased activity of GPX4 occur during ferroptosis. Inhibitions of GPX4 activity prevent the reduction reaction mediated by GPX4 from metabolizing lipid peroxides, resulting in their accumulation [48].
Iron overload is another important feature of ferroptosis. The Fenton reaction, which results in non-enzymatic lipid peroxidation, regulates ferroptosis by producing lethal reactive oxygen species (ROS) [49]. As a cofactor for iron-containing enzymes, iron may also be essential for enzymatic lipid metabolism. Therefore, iron appears to play a vital role in ferroptosis, whether enzymatically or non-enzymatically, in the production of ROS (Figure 3). In addition to the above-mentioned GPX4-mediated classical ferroptosis pathway, the mitochondrial transmembrane channel VDAC (voltage-dependent anion channel) and the tumor suppressor gene P53 can also mediate ferroptosis [50][51].
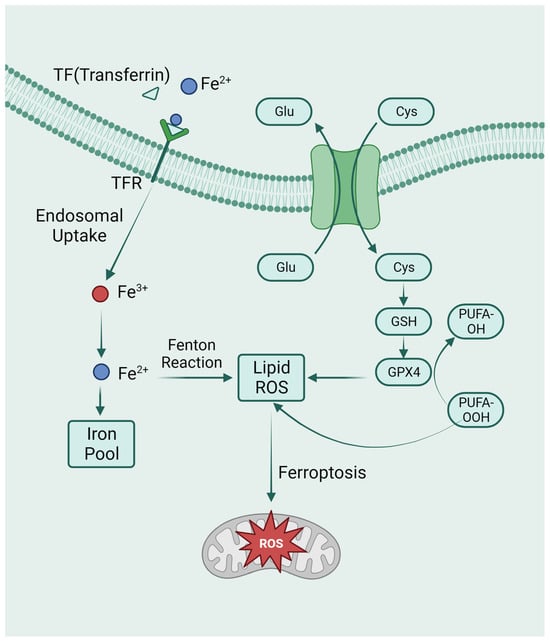
Figure 3. The metabolic pathways of ferroptosis. Intracellular glutamate (Glu) and extracellular cysteine (Cys) are transported via System Xc-, facilitating the incorporation of cysteine into glutathione (GSH) synthesis. Glutathione peroxidase 4 (GPX4) actively participates in the intracellular neutralization process, converting endogenous peroxidized polyunsaturated fatty acids (PUFAs-OOH) to their non-peroxidized counterparts (PUFAs-OH). This enzymatic activity culminates in the mitigation of reactive oxygen species (ROS) accumulation. Ferroptosis is caused by excess iron. Through TFR, circulating iron is combined with TF and enters cells. Iron in Fe3+ was deoxidized to iron in Fe2+. Ultimately, Fe2+ was released into a labile iron pool in the cytoplasm. Fenton reaction is a chain reaction between ferric ions (Fe2+) and hydrogen peroxide to catalyze the formation of OH radicals. The free Fe2+ in the iron pool participates in the Fenton reaction, generating ROS substances, and the accumulated ROS peroxidizes membrane lipids, resulting in loss of cell function and cell death.
In endometriotic lesions, erythrocyte degradation leads to iron accumulation [52][53]. Iron overload influences the preimplantation process of the endometriosis mouse embryo. Mechanically, iron overload can disrupt mitochondrial function by interfering with ATP production. Additionally, iron overload can induce intracellular ROS. Embryos cultured at higher iron concentrations showed lower rates of cleavage and blastocyst formation [54]. Treatment of mouse granulosa cells with follicular fluid from patients with endometriosis-associated infertility can induce ferroptosis, which hinders oocyte maturation by releasing exosomes [55]. These studies provide a new perspective on ferroptosis’s involvement in endometriosis-induced infertility. Some lncRNA can also regulate ferroptosis in endometriosis. For example, up-regulated ADAMTS9-AS1 (ADAM Metallopeptidase with Thrombospondin Type 1 Motif Antisense RNA 1) accelerates endometrial proliferation and migration by modulating miR-6516-5p/GPX4-dependent ferroptosis. ADAMTS9-AS1 increased ROS levels, and inhibition of this lncRNA significantly reduced GPX4 expression [56]. Ferroptosis is associated with endometriosis-derived clear cell carcinoma of the ovary (CCOC). Compared with the normal secretory endometrium, the expression of cysteine and glutathione synthesis pathway genes and the downregulation of iron antiporter were observed in CCOC [57]. According to another study, CD 10 negative endometriosis-derived mesenchymal stem cells expressed a high level of iron export proteins and were capable of transmitting iron to associated CCOC cells [58]. Significantly, the stroma may support the growth and development of tumor cells through iron transport and donation. Further characterization of the stromal phenotype may be a new direction in the study of malignant transformation in endometriosis.
One of the characteristics of endometriosis foci is myofibroblast-induced fibrosis and angiogenesis. Ferroptosis is also involved in these processes, and studies by Zhang et al. showed that ferroptosis inhibitors could reduce the proportion of myofibroblasts in endometriosis lesions and alleviate fibrosis [59]. Endometrial stromal cell ferroptosis in the ovarian endometrioma may promote angiogenesis [60]. Erastin, a ferroptosis inducer, can shrink endometriosis lesions, but the mechanism remains to be explored [61]. The MALAT1/miR-145-5p/MUC1 axis was involved in shrinking endometriotic lesions caused by erastin-induced ferroptosis [62]. Perhaps there are other regulatory mechanisms for the ameliorating effect of erastin on endometriosis, and this ferroptosis inhibitor can be applied to the drug treatment of endometriosis in the future.
4. Cuproptosis in Endometriosis
New research has revealed that copper-dependent cuproptosis is a non-apoptotic mode of cell death that regulates mitochondrial respiration. During cuproptosis, copper ions are combined with fatty acylated components in the tricarboxylic acid cycle (Figure 4). Consequently, fatty acylated proteins aggregate and iron-sulfur cluster proteins are reduced, resulting in protein toxicity stress and cell death [63]. FDX1(Ferredoxin 1) is a ferrite-reducing protein, which is the core molecule of cuproptosis. On the one hand, FDX1 can reduce Cu2+ to Cu+, which is more toxic, to induce cuproptosis. On the other hand, it can catalyze the lipacylation of pyruvate dehydrogenase core structural proteins [64]. According to a recent study, FDX1 mediates cuproptosis in endometriosis through the G6PD pathway, which inhibits the proliferation and metastasis of endometriosis cells [64]. It is still unclear how cuproptosis occurs in endometriosis, and research in this field holds great promise.
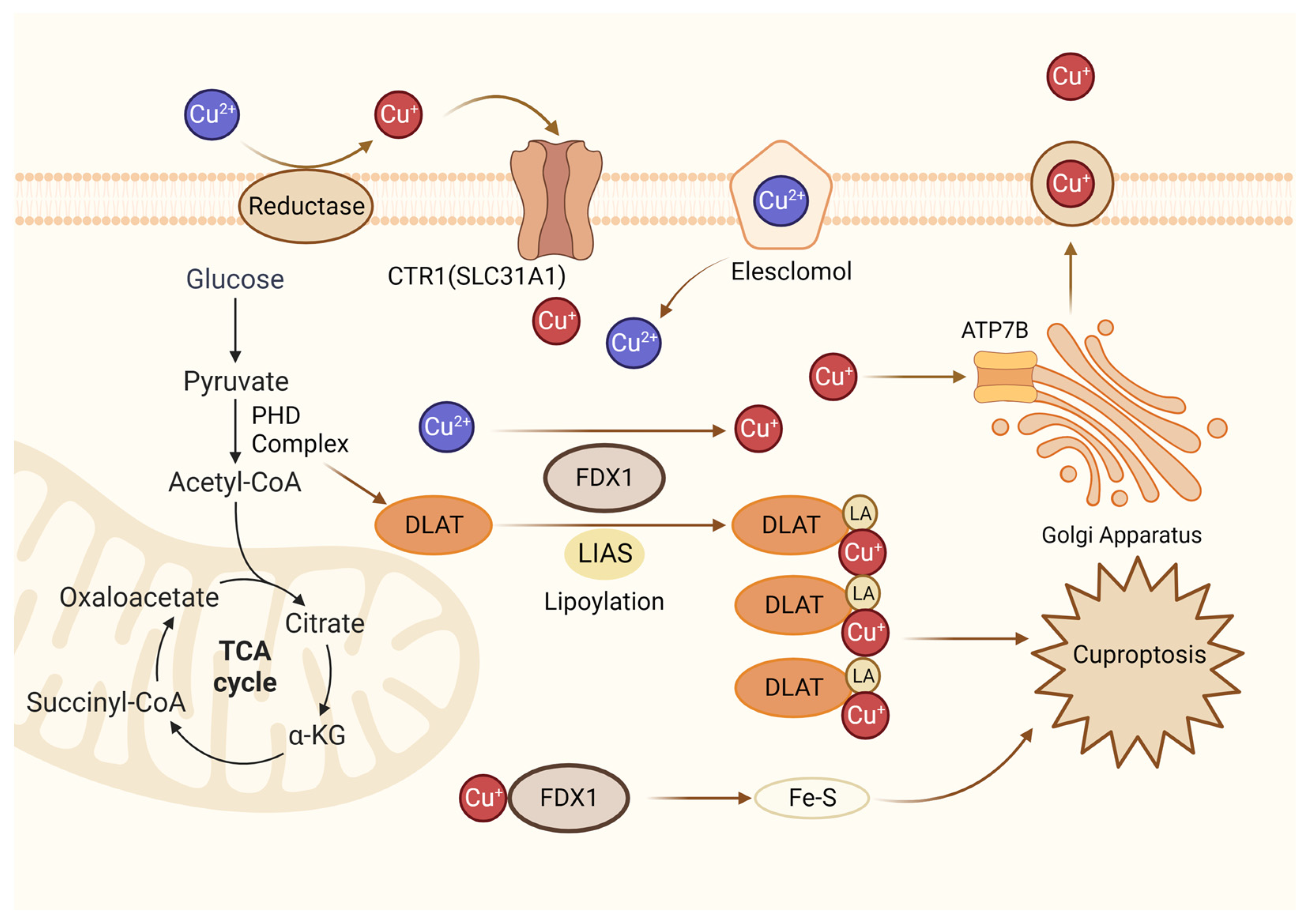
Figure 4. Diagrammatic representation of cuproptosis mechanism. Cuproptosis suppresses mitochondrial oxidative respiration through the inhibition of DLAT, a crucial constituent within the pyruvate dehydrogenase complex (PDH complex). Elesclomol binds extracellular Cu and transports it to intracellular compartments. Upon entry into the cell, Cu engages with lipoylated mitochondrial enzymes in the tricarboxylic acid cycle (TCA), such as DLAT. LIAS/FDX1 regulates protein lipoylation, facilitating mitochondrial protein aggregation and Fe-S cluster loss. As a result of these aberrant processes, proteotoxic stress occurs, and cells die. Lipoylation is a post-translational modification specific to mitochondrial proteins. LA: lipoic acid. DLAT: dihydrolipoyl transacetylase.
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257.
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537.
- Hockenbery, D. Defining apoptosis. Am. J. Pathol. 1995, 146, 16–19.
- Savill, J. Apoptosis in disease. Eur. J. Clin. Investig. 1994, 24, 715–723.
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16.
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417.
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109.
- Ren, Y.Q.; Wang, X.R.; Guo, J.Y.; Wang, D.; Li, X.H.; Cheng, X.M.; Wang, X.G. CHCHD2 Regulates Mitochondrial Function and Apoptosis of Ectopic Endometrial Stromal Cells in the Pathogenesis of Endometriosis. Reprod. Sci. 2022, 29, 2152–2164.
- Gebel, H.M.; Braun, D.P.; Tambur, A.; Frame, D.; Rana, N.; Dmowski, W.P. Spontaneous apoptosis of endometrial tissue is impaired in women with endometriosis. Fertil. Steril. 1998, 69, 1042–1047.
- Dmowski, W.P.; Gebel, H.; Braun, D.P. Decreased apoptosis and sensitivity to macrophage mediated cytolysis of endometrial cells in endometriosis. Hum. Reprod. Update 1998, 4, 696–701.
- Cullen, S.P.; Martin, S.J. Fas and TRAIL ‘death receptors’ as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 2015, 39, 26–34.
- Han, S.J.; Jung, S.Y.; Wu, S.P.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.J.; et al. Estrogen Receptor β Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974.
- Béliard, A.; Noël, A.; Foidart, J.M. Reduction of apoptosis and proliferation in endometriosis. Fertil. Steril. 2004, 82, 80–85.
- Fujino, K.; Yamashita, Y.; Hayashi, A.; Asano, M.; Morishima, S.; Ohmichi, M. Survivin gene expression in granulosa cells from infertile patients undergoing in vitro fertilization-embryo transfer. Fertil. Steril. 2008, 89, 60–65.
- Corachán, A.; Pellicer, N.; Pellicer, A.; Ferrero, H. Novel therapeutic targets to improve IVF outcomes in endometriosis patients: A review and future prospects. Hum. Reprod. Update 2021, 27, 923–972.
- Chen, L.; Ni, Z.X.; Cai, Z.L.; Cheng, W.; Sun, S.; Yu, C.Q.; JinYu. The Mechanism Exploration of Follicular Fluids on Granulose Cell Apoptosis in Endometriosis-Associated Infertility. Biomed. Res. Int. 2021, 2021, 6464686.
- Sreerangaraja Urs, D.B.; Wu, W.H.; Komrskova, K.; Postlerova, P.; Lin, Y.F.; Tzeng, C.R.; Kao, S.H. Mitochondrial Function in Modulating Human Granulosa Cell Steroidogenesis and Female Fertility. Int. J. Mol. Sci. 2020, 21, 3592.
- Liu, W.; Hu, B.; Wang, X.; Huang, E.; Chen, X.; Chen, L. GRIK1-AS1 deficiency accelerates endometriosis progression by boosting DNMT1-dependent SFRP1 promoter methylation in endometrial stromal cells. J. Gene Med. 2023, 25, e3557.
- Zhang, L.; Yu, Z.; Qu, Q.; Li, X.; Lu, X.; Zhang, H. Exosomal lncRNA HOTAIR Promotes the Progression and Angiogenesis of Endometriosis via the miR-761/HDAC1 Axis and Activation of STAT3-Mediated Inflammation. Int. J. Nanomed. 2022, 17, 1155–1170.
- Antonio, L.G.L.; Meola, J.; Rosa, E.S.A.; Nogueira, A.A.; Candido Dos Reis, F.J.; Poli-Neto, O.B.; Rosa, E.S.J.C. Altered Differential Expression of Genes and microRNAs Related to Adhesion and Apoptosis Pathways in Patients with Different Phenotypes of Endometriosis. Int. J. Mol. Sci. 2023, 24, 4434.
- Zhang, A.; Wang, G.; Jia, L.; Su, T.; Zhang, L. Exosome-mediated microRNA-138 and vascular endothelial growth factor in endometriosis through inflammation and apoptosis via the nuclear factor-κB signaling pathway. Int. J. Mol. Med. 2019, 43, 358–370.
- Park, Y.; Cho, Y.J.; Sung, N.; Park, M.J.; Guan, X.; Gibbons, W.E.; O’Malley, B.W.; Han, S.J. Oleuropein suppresses endometriosis progression and improves the fertility of mice with endometriosis. J. Biomed. Sci. 2022, 29, 100.
- Mai, H.; Liao, Y.; Luo, S.F.; Wei, K.Y.; Yang, F.; Shi, H.J. Histone deacetylase HDAC2 silencing prevents endometriosis by activating the HNF4A/ARID1A axis. J. Cell. Mol. Med. 2021, 25, 9972–9982.
- Rao, Z.; Zhu, Y.; Yang, P.; Chen, Z.; Xia, Y.; Qiao, C.; Liu, W.; Deng, H.; Li, J.; Ning, P.; et al. Pyroptosis in inflammatory diseases and cancer. Theranostics 2022, 12, 4310–4329.
- Li, Y.; Yuan, Y.; Huang, Z.X.; Chen, H.; Lan, R.; Wang, Z.; Lai, K.; Chen, H.; Chen, Z.; Zou, Z.; et al. GSDME-mediated pyroptosis promotes inflammation and fibrosis in obstructive nephropathy. Cell Death Differ. 2021, 28, 2333–2350.
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572.
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782.
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298.
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353.
- Hang, Y.; Tan, L.; Chen, Q.; Liu, Q.; Jin, Y. E3 ubiquitin ligase TRIM24 deficiency promotes NLRP3/caspase-1/IL-1β-mediated pyroptosis in endometriosis. Cell Biol. Int. 2021, 45, 1561–1570.
- Kelleher, A.M.; Peng, W.; Pru, J.K.; Pru, C.A.; DeMayo, F.J.; Spencer, T.E. Forkhead box a2 (FOXA2) is essential for uterine function and fertility. Proc. Natl. Acad. Sci. USA 2017, 114, E1018–E1026.
- Feng, Y.; Tan, B.; Dong, H.; Zheng, L. FoxA2 represses ERβ-mediated pyroptosis in endometriosis by transcriptionally inhibiting IGF2BP1. Exp. Cell Res. 2023, 426, 113539.
- Huang, Y.; Li, R.; Hu, R.; Yao, J.; Yang, Y. PEG2-Induced Pyroptosis Regulates the Expression of HMGB1 and Promotes hEM15A Migration in Endometriosis. Int. J. Mol. Sci. 2022, 23, 11707.
- Sun, J.; Gan, L.; Sun, J. Identification and Validation of Three m6A Regulators: FTO, HNRNPC, and HNRNPA2B1 as Potential Biomarkers for Endometriosis. Genes 2022, 14, 86.
- Guo, Q.; Zhou, C.; Xiang, Y.; Liang, X. Pyroptosis orchestrates immune responses in endometriosis. Int. Immunopharmacol. 2023, 118, 110141.
- Samimi, M.; Pourhanifeh, M.H.; Mehdizadehkashi, A.; Eftekhar, T.; Asemi, Z. The role of inflammation, oxidative stress, angiogenesis, and apoptosis in the pathophysiology of endometriosis: Basic science and new insights based on gene expression. J. Cell Physiol. 2019, 234, 19384–19392.
- Nanda, A.; K, T.; Banerjee, P.; Dutta, M.; Wangdi, T.; Sharma, P.; Chaudhury, K.; Jana, S.K. Cytokines, Angiogenesis, and Extracellular Matrix Degradation are Augmented by Oxidative Stress in Endometriosis. Ann. Lab. Med. 2020, 40, 390–397.
- Laschke, M.W.; Menger, M.D. In vitro and in vivo approaches to study angiogenesis in the pathophysiology and therapy of endometriosis. Hum. Reprod. Update 2007, 13, 331–342.
- Zhang, M.; Shi, Z.; Peng, X.; Cai, D.; Peng, R.; Lin, Y.; Dai, L.; Li, J.; Chen, Y.; Xiao, J.; et al. NLRP3 inflammasome-mediated Pyroptosis induce Notch signal activation in endometriosis angiogenesis. Mol. Cell Endocrinol. 2023, 574, 111952.
- Xu, Y.; Liu, H.; Xiong, W.; Peng, Y.; Li, X.; Long, X.; Jin, J.; Liang, J.; Weng, R.; Liu, J.; et al. A novel mechanism regulating pyroptosis-induced fibrosis in endometriosis via lnc-MALAT1/miR-141-3p/NLRP3 pathway†. Biol. Reprod. 2023, 109, 156–171.
- Yang, F.; Xiao, Y.; Ding, J.H.; Jin, X.; Ma, D.; Li, D.Q.; Shi, J.X.; Huang, W.; Wang, Y.P.; Jiang, Y.Z.; et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023, 35, 84–100.e108.
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125.
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227.
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System X(c)(-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292.
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020, 27, 662–675.
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692.
- von Krusenstiern, A.N.; Robson, R.N.; Qian, N.; Qiu, B.; Hu, F.; Reznik, E.; Smith, N.; Zandkarimi, F.; Estes, V.M.; Dupont, M.; et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol. 2023, 19, 719–730.
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822.
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62.
- Niu, B.; Lei, X.; Xu, Q.; Ju, Y.; Xu, D.; Mao, L.; Li, J.; Zheng, Y.; Sun, N.; Zhang, X.; et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol. Toxicol. 2022, 38, 505–530.
- Wyatt, J.; Fernando, S.M.; Powell, S.G.; Hill, C.J.; Arshad, I.; Probert, C.; Ahmed, S.; Hapangama, D.K. The role of iron in the pathogenesis of endometriosis: A systematic review. Hum. Reprod. Open 2023, 2023, hoad033.
- Van Langendonckt, A.; Casanas-Roux, F.; Donnez, J. Iron overload in the peritoneal cavity of women with pelvic endometriosis. Fertil. Steril. 2002, 78, 712–718.
- Chen, X.; Zhou, Y.; Wu, D.; Shu, C.; Wu, R.; Li, S.; Huang, Q.; Shu, J. Iron overload compromises preimplantation mouse embryo development. Reprod. Toxicol. 2021, 105, 156–165.
- Ni, Z.; Li, Y.; Song, D.; Ding, J.; Mei, S.; Sun, S.; Cheng, W.; Yu, J.; Zhou, L.; Kuang, Y.; et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. 2022, 13, 579.
- Wan, Y.; Gu, C.; Kong, J.; Sui, J.; Zuo, L.; Song, Y.; Chen, J. Long noncoding RNA ADAMTS9-AS1 represses ferroptosis of endometrial stromal cells by regulating the miR-6516-5p/GPX4 axis in endometriosis. Sci. Rep. 2022, 12, 2618.
- Beddows, I.; Fan, H.; Heinze, K.; Johnson, B.K.; Leonova, A.; Senz, J.; Djirackor, S.; Cho, K.R.; Pearce, C.L.; Huntsman, D.G.; et al. Cell state of origin impacts development of distinct endometriosis-related ovarian carcinoma histotypes. Cancer Res. 2023, 84, 26–38.
- Atiya, H.I.; Frisbie, L.; Goldfeld, E.; Orellana, T.; Donnellan, N.; Modugno, F.; Calderon, M.; Watkins, S.; Zhang, R.; Elishaev, E.; et al. Endometriosis-Associated Mesenchymal Stem Cells Support Ovarian Clear Cell Carcinoma through Iron Regulation. Cancer Res. 2022, 82, 4680–4693.
- Zhang, Y.; Liu, X.; Deng, M.; Xu, C.; Zhang, Y.; Wu, D.; Tang, F.; Yang, R.; Miao, J. Ferroptosis induced by iron overload promotes fibrosis in ovarian endometriosis and is related to subpopulations of endometrial stromal cells. Front. Pharmacol. 2022, 13, 930614.
- Li, G.; Lin, Y.; Zhang, Y.; Gu, N.; Yang, B.; Shan, S.; Liu, N.; Ouyang, J.; Yang, Y.; Sun, F.; et al. Endometrial stromal cell ferroptosis promotes angiogenesis in endometriosis. Cell Death Discov. 2022, 8, 29.
- Li, Y.; Zeng, X.; Lu, D.; Yin, M.; Shan, M.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964.
- Liang, Z.; Wu, Q.; Wang, H.; Tan, J.; Wang, H.; Gou, Y.; Cao, Y.; Li, Z.; Zhang, Z. Silencing of lncRNA MALAT1 facilitates erastin-induced ferroptosis in endometriosis through miR-145-5p/MUC1 signaling. Cell Death Discov. 2022, 8, 190.
- Wang, D.; Tian, Z.; Zhang, P.; Zhen, L.; Meng, Q.; Sun, B.; Xu, X.; Jia, T.; Li, S. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomed. Pharmacother. 2023, 163, 114830.
- Lu, J.; Ling, X.; Sun, Y.; Liu, L.; Liu, L.; Wang, X.; Lu, C.; Ren, C.; Han, X.; Yu, Z. FDX1 enhances endometriosis cell cuproptosis via G6PD-mediated redox homeostasis. Apoptosis 2023, 28, 1128–1140.
More
Information
Subjects:
Obstetrics & Gynaecology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
787
Revisions:
2 times
(View History)
Update Date:
17 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No