Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ewa Robak | -- | 2506 | 2024-02-07 20:43:37 | | | |
| 2 | Sirius Huang | Meta information modification | 2506 | 2024-02-08 03:03:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Stein, T.; Robak, T.; Biernat, W.; Robak, E. Primary Cutaneous CD30-Positive Lymphoproliferative Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/54872 (accessed on 25 June 2026).
Stein T, Robak T, Biernat W, Robak E. Primary Cutaneous CD30-Positive Lymphoproliferative Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/54872. Accessed June 25, 2026.
Stein, Tomasz, Tadeusz Robak, Wojciech Biernat, Ewa Robak. "Primary Cutaneous CD30-Positive Lymphoproliferative Disorders" Encyclopedia, https://encyclopedia.pub/entry/54872 (accessed June 25, 2026).
Stein, T., Robak, T., Biernat, W., & Robak, E. (2024, February 07). Primary Cutaneous CD30-Positive Lymphoproliferative Disorders. In Encyclopedia. https://encyclopedia.pub/entry/54872
Stein, Tomasz, et al. "Primary Cutaneous CD30-Positive Lymphoproliferative Disorders." Encyclopedia. Web. 07 February, 2024.
Copy Citation
One of the most common subgroups of cutaneous T-cell lymphomas is that of primary cutaneous CD30-positive lymphoproliferative disorders. The group includes lymphomatoid papulosis (LyP) and primary cutaneous anaplastic large cell lymphoma (pcALCL), as well as some borderline cases. Significant progress has been made in understanding the genetics and treatment of these disorders.
anaplastic large cell lymphoma
brentuximab vedotin
CD30-positive lymphomas
diagnosis
lymphomatoid papulosis
primary cutaneous anaplastic large cell lymphoma
1. Introduction
Primary cutaneous CD30+ lymphoproliferative disorders (LPDs) are the second most common group of primary cutaneous T-cell lymphomas, after mycosis fungoides (MFs) [1]. The group includes primary cutaneous anaplastic large cell lymphoma (pcALCL), lymphomatoid papulosis (LyP) and borderline tumours. They generally have a good prognosis, but most cases are also characterised by a long-term and recurrent course that makes everyday life difficult for patients. It is worth emphasising that although these are indolent proliferations, they may pose a risk of internal organ involvement. In addition, although LyP is a benign disease, it is still included in the group of lymphomas because, in rare cases, it can transform from LyP to MF, pcALCL or Hodgkin lymphoma (HL). Generally, LPD30+ disorders have a slow and chronic course. Currently, brentuximab vedotin (BV) is one of the newer drugs used to treat CD30+ LPD [2]. Brentuximab vedotin is a conjugate of a mouse–human chimeric IgG1 anti-CD30 antibody linked to monomethyl auristatin E (MMAE), an anti-tubulin drug. Treatment brings the rapid remission of skin lesions but also causes side effects, the most common of which is peripheral neuropathy. It is believed that the neuropathy that can develop during brentuximab vedotin therapy is caused by the inhibition of microtubule-dependent axonal transport.
CD30, also known as TNFRSF8 (TNF Receptor Superfamily Member 8) or Ki-1, is a 120 kDa glycoprotein receptor belonging to the TNF family [3]. It was first identified in 1982 as a surface marker for selected Hodgkin lymphoma (HL) cell lines [4]. The CD30 receptor is activated by the binding of its ligand, CD30L, a membrane-bound cytokine expressed on activated granulocytes and lymphocytes [5]. CD30 is most highly expressed in CD8+ and CD4+ lymphocytes [6]. The formation of the ligand complex activates the receptor and TNF-related factor recruitment (TRAF1, TRAF2, and TRAF3); it also initiates the binding of proteins to form a signalling complex inside the cell. This stimulates the nuclear factor kappa B (NFkB) pathway and activates signalling via the mitogen-activated protein kinase (MAPK) pathways. Both pathways promote a variety of effects, including those that promote survival and prevent apoptosis of neoplastic cells. Moreover, it is postulated that CD30 activation is regulated via the expression of Jun-B, which is a transcription factor of activated proteins (AP-1) responsible for neoplastic transformation [7].
In healthy individuals, CD30+ expression is minimal, most prominent in activated lymphocytes; even in this case, it only accounts for less than 1% of activated lymphocytes. In healthy individuals, the role of CD30 is thought to supervise the immune system by mediating information between B and T cells.
Importantly, CD30 expression is also observed on CD8+ and CD4+ lymphocytes in skin inflammation, viral infections and malignancies. Increased amounts of CD30 have been found in atopic dermatitis, psoriasis, parasitic infections (scabies) [8], as well as in viral infections with Epstein–Barr virus (EBV), human immunodeficiency virus (HIV), human T-lymphoma virus 1 (HTLV-1) and molluscum contagiosum [9]. Falini et al. showed that the viral infection could increase the number of activated CD30-expressing cells from 0.1% to 95% within three days [10].
CD30 expression is characteristic of LyP and pcALCL; however, it is not unique to primary cutaneous CD30+ LPD and cannot be defined as a marker of these disorders. It is also found in anaplastic large cell lymphoma (ALCL), HL, large cell transformation of mycosis fungoides (MF-LCT), acute myeloid leukaemia (AML), myelodysplastic syndromes (MDS), mastocytosis, CD30+ B-cell lymphomas and EBV+ hydroa vacciniforme-like-T-cell lymphoma [11]. CD30 expression varies across the six WHO-recognised LyP histological subtypes. Primary cutaneous anaplastic large cell lymphoma cells express CD30 at 75%.
In clinical practice, CD30 expression on the cell surface can be assessed by flow cytometry (FCM) and immunohistochemistry (IHC) [9]. The most common evaluation method is the immunohistochemistry of skin specimens, which can be combined with an assessment of cell morphology. There are many different anti-CD30 antibodies that recognise epitopes on CD30, but the BerH2 antibody is used for the routine IHC assessment of CD30 [5]. Given recent molecular discoveries, therapies targeting the CD30 receptor appear attractive to researchers trying to identify targeted therapies for these lymphomas.
2. Primary Cutaneous CD30-Positive Lymphoproliferative Disorders
According to the current consensus of the fifth edition of the classification of cutaneous lymphomas of the World Health Organization (WHO) and the European Organization for Research and Treatment of Cancer (EORTC), primary cutaneous CD30+ LPD constitutes a separate group of lymphomas [1][12]. They are the second most common group of CTCLs, accounting for approximately 25% [13][14].
Primary cutaneous CD30+ LPDs include pcALCL and LyP, as well as borderline tumours with clinical and histological features lying between the two. Importantly, as the histological criteria are often not sufficient to distinguish between these diseases, researchers typically use the term CD30+ LPD during the initial clinical evaluation, especially pathological diagnosis, rather than LyP or pcALCL. A short follow-up period (8–10 weeks) may reveal spontaneous regression, which is more characteristic of LyP. This diagnosis is critical for further disease management and the initiation of appropriate therapy. Both LyP and pcALCL have different histological, clinical and immunophenotypic variants. Also, despite their histological picture suggesting highly malignant neoplastic infiltration, they also generally have a slow course with a good prognosis [15].
2.1. Lymphomatoid Papulosis
Lymphomatoid papulosis was first described by Dr. Warren L. Macauley in 1968 as a benign, histologically malignant, self-limiting, but recurrent disorder of unknown aetiology [16]. It was only after years of follow-up that biopsies from skin lesions demonstrated a histology typical of lymphoma together with the presence of large atypical CD30-positive cells; as such, LyP was classified as a CD30+ LPD [17].
Lymphomatoid papulosis is a rare skin proliferation with an incidence of 1.2–1.9/million and an excellent prognosis, with a 10-year survival rate of approximately 100% [8]. However, patients with LyP are at risk of developing secondary malignancies, including nodal or cutaneous ALCL, HL and MF. These lymphomas are clonally related to LyP and, according to various sources, develop in 4 to 60% of LyP patients. They can occur before, concomitantly or after LyP [8][18][19]. The most common secondary lymphomas identified in a large retrospective cohort study of 180 patients with LyP were MF (61.4%) and ALCL (26.3%) [20]. Sauder et al. report that LyP B or C, male sex, LyP with monoclonal rearrangement of the TCR receptor, EBV infection and advanced age increase the chance of second malignancy; as such, each patient diagnosed with this disease requires increased oncological supervision [13].
The pathogenesis of LyP is unknown, but most studies suggest a genetic background based on abnormalities in the CD30 transcription [21]. Although viral infections with HTLV-1 virus and hepatitis E virus were also suspected, these correlations were not confirmed in subsequent studies [22][23]. It has been suggested that LyP may be related to iatrogenic inflammation of the skin: Haro et al. [24] found that a patient previously treated with radiotherapy for breast cancer later developed LyP in the treated area.
Lymphomatoid papulosis manifests as polymorphic, varicella-like papules, sometimes vesicles, necrotic, ulcerated or haemorrhagic lesions [21] (Figure 1). They are most common on the limbs and trunk; however, in some cases, they are also found on the genitals and oral cavity. The lesions may be accompanied by itching or slight tenderness or may be asymptomatic. The main feature of Lyp that distinguishes it from other types of CD30+ LPD is that the lesions spontaneously resolve within four to eight weeks, in which case, resolution may be permanent or a few years [25]. The disorder occurs in adults, with a slight predominance of men aged 20 to 40 years, but paediatric cases have also been reported [21]. The WHO-EORTS 2018 update classifies LyP into six subtypes, viz. LyP A, B, C, D, E and LyP with DUSP22-IRF4 rearrangement; these differ slightly in histopathology and immunophenotypes, but some features overlap [13].
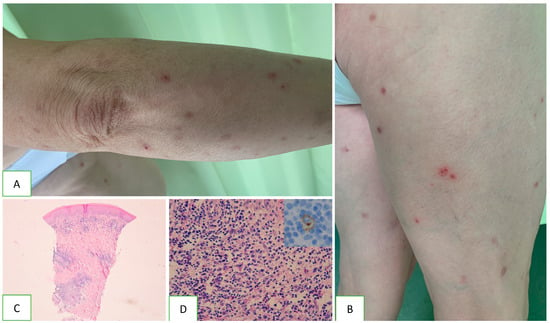
Figure 1. Clinical characteristics and histopathology of lymphoid papulosis. Scattered papules and single nodules are visible on the skin of the limbs and trunk (A). Some lesions have a haemorrhagic component, and others present disintegrations in the centre (B). Typical arrangement of the cutaneous infiltrate with nodular aggregates sparing the epidermis (C). Cellular composition contains small lymphocytes and scattered eosinophils with single atypical CD30+ cells (D).
The most common subtype is LyP A > 80%. All subtypes have an indolent clinical course and similar clinical presentation, with the exception of subtype E; this subtype manifests as vasculitis involving small and medium vessels with large ulcerative lesions [26]. The immunophenotype of the LyP cells varies by subtype. They are generally represented by CD4+CD8− or CD4−CD8+, but CD4+CD8+ cases are rare. LyP with DUSP22 is more commonly CD4−/CD8−. Variable loss of pan T cell antigens (CD2, CD3, CD5 and CD7) can be observed. Most cases are CD30+, BF1+, granzyme B+ and CD56+/−. Clonal rearrangement of T-cell receptor (TCR) genes has been reported in more than 50% of cases of LyP. The majority of LyP cases express the alpha/beta TCR. However, expression of the gamma/delta TCR has been observed in type D [15].
The diagnosis of LyP is based on a combination of clinical features and histopathological and immunohistochemical findings [21]. It is necessary to perform a complete blood count, LDH determination and basic blood biochemistry. If a secondary malignancy is suspected, e.g., the presence of enlarged lymph nodes, B symptoms or skin lesions not resolving spontaneously, the diagnostics should be extended to include further laboratory and imaging tests, possibly in combination with a diagnostic with further laboratory and imaging tests, and maybe with bone marrow biopsy [1].
2.2. Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous anaplastic CD30+ cell lymphoma belongs to the subgroup of peripheral T-cell lymphomas (PTCL). It was described in 1985 as a CD30-expressing malignancy of the lymphatic system, which, in most cases, affects only the skin [27][28]. It contains large cells with pleomorphic, immunoblastic or anaplastic morphology (Figure 2). The CD30 antigen can be found in at least 75% of neoplastic cells [19][29]. The condition accounts for 9% of all CTCLs. It usually affects the elderly (median 60 years), with a greater prevalence among men, but has also been reported in children [18]. The 10-year survival rate for pcALCL is 95%, while the 5-year survival rate is 93% for ALK-positive ALCL and 37% for ALK-negative ALCL [25][30].
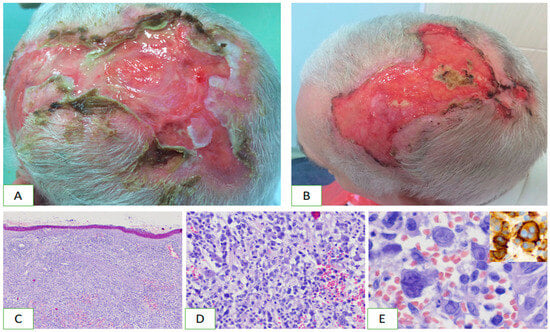
Figure 2. Clinical characteristics and histopathology of primary cutaneous anaplastic large cell lymphoma. On the top of the head, there are extensive merging ulcers accompanied by pain (A,B). Diffuse infiltration of lymphoma cells involves the skin (C). It contains atypical pleomorphic cells (D). Mononucleated and multinucleated cells prevail and show strong CD30 expression (E).
Primary cutaneous anaplastic large cell lymphoma is clinically manifested as solitary or multiple nodules, tumours, erythematous plaques and sometimes papular lesions, often accompanied by ulceration (Figure 2). They tend to grow bigger over weeks and usually involve the head, neck and limbs. Extensive limb disease (ELD), a variant of pcALCL, is manifested by multiple skin tumours on one limb and is associated with disease progression and poorer prognosis [31]. Additionally, pcALCL is less likely to demonstrate spontaneous regression than LyP, ranging from 10% to 42% of patients, and is often subject to recurrences [32]. Extracutaneous involvement is rare and most commonly involves regional lymph nodes, which are observed in 10% of patients [25]. The secondary involvement of the skin by systemic ALCL, HL and MF CD30+ should be excluded.
The classic histological picture presents limited cellular infiltration and large lymphoid cells, usually with absent or discrete epidermotropism. The immunophenotype includes CD4+, CD30+, BF1+, CD56+/−, granzyme B+ cells, and variable loss of CD2, CD3 and CD5. Unlike nodal ALCLs, most pcALCLs express CLA cutaneous lymphocyte-associated antigen) but do not express EMA (epithelial membrane (antigen) [33][34].
As in the case of LyP, the diagnosis of pcALCL is based on a combination of clinical features and histopathological and immunopathological findings. Basic laboratory tests are necessary, as in LyP; however, these should be accompanied by imaging of the whole body, preferably with PET-CT. Additionally, it is worth testing for HIV, HTLV-1 and EBV because some T-cell lymphomas may produce secondary skin lesions and have a viral aetiology. If the lymph nodes are enlarged or metabolically active in PET, a lymph node biopsy should be performed. Bone marrow biopsy is no longer recommended in patients with pcALCL unless the patient has systemic symptoms, cytopenia or extensive skin lesions [15][18].
3. Genetics of Primary Cutaneous CD30-Positive Lymphoproliferative Disorders
Molecular tests ease the diagnosis of cutaneous CD30+ lymphoproliferative disorders and help differentiate LyP and pcALCL. It is also important to differentiate pcALCL and secondary skin involvement by anaplastic lymphoma kinase (ALK)-positive and ALK-negative ALCL: the latter requires different treatment methods and entails a worse prognosis. ALK is a tyrosine kinase receptor. Its physiological expression is limited to a few cell types, such as endothelial cells or glial cells. ALK fusion has been noted in pcALCL but not LyP. Until recently, pcALCL has been considered an ALK-negative lymphoma, although pcALCL may also be ALK-positive [35]; this is a rare phenomenon which occurs practically only in the paediatric population and is associated with progression to the secondary systemic involvement [36].
A new LyP subtype was recently identified, characterised by a rearrangement of DUSP22 and IRF4 (Interferon Regulatory Factor 4), a tumour suppressor gene regulating T-cell proliferation, at the 6p25.3 locus; the same rearrangements have been previously reported in pcALCL, where they are observed in 20–57% of cases [37]. DUSP22—IRF4 gene rearrangement has also been described in systemic ALCL and transformed MF [38].
Another study identified chromosomal translocations targeting CD30+ LPD tyrosine kinases. Among the 47 patients with LPD, 4% carried the NPM1 (5q35) and TYK2 (19p13) fusion, which encodes the NPM1-TYK2 protein. This protein promotes cell proliferation by activating the STAT1/3/5 pathway; this information may be valuable when using tyrosine kinase inhibitors in patients carrying this fusion [35].
Sun et al. found that in most LPDs, the SATB1 protein (binding AT1-rich sequences), i.e., a nuclear protein of thymocytes that plays a role in T cell development, is overexpressed in CD30+ lymphocytes [39]. Overexpression has been noted in 91.7% of LyP and 38.1% of pcALCL cases, with the prevalence increasing as the disease progresses. Interestingly, these cases have a better response to methotrexate (MTX).
Epigenetics can also be used to differentiate between CD30 neoplastic proliferation and CD30+ inflammatory infiltrates. De Souza et al. evaluated the expression of intracellular 5-hydroxymethylcytosine (5-hmC) caused by DNA cytosine methylation at position 5 [40]. This phenomenon occurs in several malignancies. The authors report that 5-hmC expression occurred in 27 LyP and 14 pcALCL cases and in 19 CD30+ inflammatory infiltrates. In contrast, a complete loss of 5-hmC expression was observed in 63% of LyP and 53% of pcALCL cases; such a lack of expression was a hallmark of CD30+ LPD, and this may help to distinguish neoplastic diseases from CD30+ inflammatory infiltrates [40].
Finally, Kamstrup et al. demonstrated Notch expression in 12 LyP and in 11 pcALCL cases; the transmembrane Notch receptor influences T lymphocyte proliferation and demonstrates strong expression in pcALCL lymphocytes but lower in LyP cells [41]. This discovery may determine the future use of targeted therapy with Notch antagonists and may help to differentiate LyP from pcALCL.
References
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714.
- Yi, J.H.; Kim, S.J.; Kim, W.S. Brentuximab vedotin: Clinical updates and practical guidance. Blood Res. 2017, 52, 243–253.
- Stein, H.; Foss, H.D.; Dürkop, H.; Marafioti, T.; Delsol, G.; Pulford, K.; Pileri, S.; Falini, B. CD30(+) anaplastic large cell lymphoma: A review of its histopathologic, genetic, and clinical features. Blood 2000, 96, 3681–3695.
- Anderson, J.R.; Armitage, J.O.; Weisenburger, D.D. Epidemiology of the non-Hodgkin’s lymphomas: Distributions of the major subtypes differ by geographic locations. Ann. Oncol. 1998, 9, 717–720.
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603.
- Zheng, B.; Fiumara, P.; Li, Y.V.; Georgakis, G.; Snell, V.; Younes, M.; Vauthey, J.N.; Carbone, A.; Younes, A. MEK/ERK pathway is aberrantly active in Hodgkin disease: A signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood 2003, 102, 1019–1027.
- Garces de Los Fayos Alonso, I.; Liang, H.C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93.
- Di Raimondo, C.; Parekh, V.; Song, J.Y.; Rosen, S.T.; Querfeld, C.; Zain, J.; Martinez, X.U.; Abdulla, F.R. Primary Cutaneous CD30+ Lymphoproliferative Disorders: A Comprehensive Review. Curr. Hematol. Malign Rep. 2020, 15, 333–342.
- Karube, K.; Kakimoto, Y.; Tonozuka, Y.; Ohshima, K. The expression of CD30 and its clinico-pathologic significance in peripheral T-cell lymphomas. Expert Rev. Hematol. 2021, 14, 777–787.
- Falini, B.; Pileri, S.; Pizzolo, G.; Dürkop, H.; Flenghi, L.; Stirpe, F.; Martelli, M.F.; Stein, H. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995, 85, 1–14.
- Zheng, W.; Medeiros, L.J.; Hu, Y.; Powers, L.; Cortes, J.E.; Ravandi-Kashani, F.; Kantarjian, H.H.; Wang, S.A. CD30 expression in high-risk acute myeloid leukemia and myelodysplastic syndromes. Clin. Lymphoma Myeloma Leuk. 2013, 13, 307–314.
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748.
- Sauder, M.B.; O’Malley, J.T.; LeBoeuf, N.R. CD30+ Lymphoproliferative Disorders of the Skin. Hematol. Clin. N. Am. 2017, 31, 317–334.
- Kempf, W.; Zimmermann, A.K.; Mitteldorf, C. Cutaneous lymphomas—An update 2019. Hematol. Oncol. 2019, 37, 43–47.
- Nikolaenko, L.; Zain, J.; Rosen, S.T.; Querfeld, C. CD30-Positive Lymphoproliferative Disorders. Cancer Treat. Res. 2019, 176, 249–268.
- Macaulay, W.L. Lymphomatoid papulosis. A continuing self-healing eruption, clinically benign--histologically malignant. Arch. Dermatol. 1968, 97, 23–30.
- Kempf, W. CD30+ lymphoproliferative disorders: Histopathology, differential diagnosis, new variants, and simulators. J. Cutan. Pathol. 2006, 33, 58–70.
- Chen, C.; Gu, Y.D.; Geskin, L.J. A Review of Primary Cutaneous CD30+ Lymphoproliferative Disorders. Hematol. Clin. N. Am. 2019, 33, 121–134.
- Prieto-Torres, L.; Rodriguez-Pinilla, S.M.; Onaindia, A.; Ara, M.; Requena, L.; Piris, M. CD30-positive primary cutaneous lymphoproliferative disorders: Molecular alterations and targeted therapies. Haematologica 2019, 104, 226–235.
- Wieser, I.; Oh, C.W.; Talpur, R.; Duvic, M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J. Am. Acad. Dermatol. 2016, 74, 59–67.
- Nowicka, D.; Mertowska, P.; Mertowski, S.; Hymos, A.; Forma, A.; Michalski, A.; Morawska, I.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E. Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System. Cells 2022, 11, 3697.
- Namba, H.; Hamada, T.; Iwatsuki, K. Human T-cell leukemia virus type 1-positive lymphomatoid papulosis. Eur. J. Dermatol. 2016, 26, 194–195.
- Mallet, V.; Bruneau, J.; Zuber, J.; Alanio, C.; Leclerc-Mercier, S.; Roque-Afonso, A.M.; Kraft, A.R.M.; Couronné, L.; Roulot, D.; Wedemeyer, H.; et al. Hepatitis E virus-induced primary cutaneous CD30(+) T cell lymphoproliferative disorder. J. Hepatol. 2017, 67, 1334–1339.
- Haro, R.; Juarez, A.; Díaz, J.L.; Santonja, C.; Manzarbeitia, F.; Requena, L. Regional lymphomatoid papulosis of the breast restricted to an area of prior radiotherapy. Cutis 2016, 97, E15–E19.
- Bekkenk, M.W.; Geelen, F.A.; van Voorst Vader, P.C.; Heule, F.; Geerts, M.L.; van Vloten, W.A.; Meijer, C.J.; Willemze, R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: A report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000, 95, 3653–3661.
- Kempf, W.; Kazakov, D.V.; Schärer, L.; Rütten, A.; Mentzel, T.; Paredes, B.E.; Palmedo, G.; Panizzon, R.G.; Kutzner, H. Angioinvasive lymphomatoid papulosis: A new variant simulating aggressive lymphomas. Am. J. Surg. Pathol. 2013, 37, 1–13.
- Kempf, W.; Pfaltz, K.; Vermeer, M.H.; Cozzio, A.; Ortiz-Romero, P.L.; Bagot, M.; Olsen, E.; Kim, Y.H.; Dummer, R.; Pimpinelli, N.; et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: Lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 2011, 118, 4024–4035.
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858.
- Saleh, J.S.; Subtil, A.; Hristov, A.C. Primary cutaneous T-cell lymphoma: A review of the most common entities with focus on recent updates. Hum. Pathol. 2023, 138, 76–102.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- Woo, D.K.; Jones, C.R.; Vanoli-Storz, M.N.; Kohler, S.; Reddy, S.; Advani, R.; Hoppe, R.T.; Kim, Y.H. Prognostic factors in primary cutaneous anaplastic large cell lymphoma: Characterization of clinical subset with worse outcome. Arch. Dermatol. 2009, 145, 667–674.
- Vu, K.; Ai, W. Update on the Treatment of Anaplastic Large Cell Lymphoma. Curr. Hematol. Malign Rep. 2018, 13, 135–141.
- Benner, M.F.; Willemze, R. Applicability and prognostic value of the new TNM classification system in 135 patients with primary cutaneous anaplastic large cell lymphoma. Arch. Dermatol. 2009, 145, 1399–1404.
- Kempf, W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin Diagn Pathol. 2017, 34, 22–35.
- Querfeld, C.; Khan, I.; Mahon, B.; Nelson, B.P.; Rosen, S.T.; Evens, A.M. Primary cutaneous and systemic anaplastic large cell lymphoma: Clinicopathologic aspects and therapeutic options. Oncology 2010, 24, 574–587.
- Pulitzer, M.; Ogunrinade, O.; Lin, O.; Steinherz, P. ALK-positive (2p23 rearranged) anaplastic large cell lymphoma with localization to the skin in a pediatric patient. J. Cutan. Pathol. 2015, 42, 182–187.
- Karai, L.J.; Kadin, M.E.; Hsi, E.D.; Sluzevich, J.C.; Ketterling, R.P.; Knudson, R.A.; Feldman, A.L. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am. J. Surg. Pathol. 2013, 37, 1173–1181.
- Runge, J.S.; Novice, M.L.; Briones, N.; Williams, K.; Lowe, L.; Boyer, D.F.; Wilcox, R.A.; Tejasvi, T.; Hristov, A.C. Patch/plaque mycosis-fungoides-like presentations of DUSP22-translocated T-cell lymphomas. J. Cutan. Pathol. 2022, 49, 299–305.
- Sun, J.; Yi, S.; Qiu, L.; Fu, W.; Wang, A.; Liu, F.; Wang, L.; Wang, T.; Chen, H.; Wang, L.; et al. SATB1 defines a subtype of cutaneous CD30+ lymphoproliferative disorders associated with a T-helper 17 cytokine profile. J. Investig. Dermatol. 2018, 138, 1795–1804.
- De Souza, A.; Tinguely, M.; Pfaltz, M.; Burghart, D.R.; Kempf, W. Loss of expression of 5-hydroxymethylcytosine in CD30-positive cutaneous lymphoproliferative disorders. J. Cutan. Pathol. 2014, 41, 901–906.
- Kamstrup, M.R.; Ralfkiaer, E.; Skovgaard, G.L.; Gniadecki, R. Potential involvement of Notch1 signalling in the pathogenesis of primary cutaneous CD30-positive lymphoproliferative disorders. Br. J. Dermatol. 2008, 158, 747–753.
More
Information
Subjects:
Dermatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
680
Revisions:
2 times
(View History)
Update Date:
08 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No