Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ludmila Zylinska | -- | 3681 | 2024-02-07 05:22:18 | | | |
| 2 | Lindsay Dong | Meta information modification | 3681 | 2024-02-08 08:34:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lisek, M.; Tomczak, J.; Boczek, T.; Zylinska, L. Calcium-Associated Proteins in Neuroregeneration. Encyclopedia. Available online: https://encyclopedia.pub/entry/54827 (accessed on 29 June 2026).
Lisek M, Tomczak J, Boczek T, Zylinska L. Calcium-Associated Proteins in Neuroregeneration. Encyclopedia. Available at: https://encyclopedia.pub/entry/54827. Accessed June 29, 2026.
Lisek, Malwina, Julia Tomczak, Tomasz Boczek, Ludmila Zylinska. "Calcium-Associated Proteins in Neuroregeneration" Encyclopedia, https://encyclopedia.pub/entry/54827 (accessed June 29, 2026).
Lisek, M., Tomczak, J., Boczek, T., & Zylinska, L. (2024, February 07). Calcium-Associated Proteins in Neuroregeneration. In Encyclopedia. https://encyclopedia.pub/entry/54827
Lisek, Malwina, et al. "Calcium-Associated Proteins in Neuroregeneration." Encyclopedia. Web. 07 February, 2024.
Copy Citation
The dysregulation of intracellular calcium levels is a critical factor in neurodegeneration, leading to the aberrant activation of calcium-dependent processes and, ultimately, cell death. Ca2+ signals vary in magnitude, duration, and the type of neuron affected. A moderate Ca2+ concentration can initiate certain cellular repair pathways and promote neuroregeneration.
calcium
neuroregeneration
CaM kinase II
oncomodulin
caldendrin
calneuron
1. Introduction
Traumatic brain injuries and neurodegenerative diseases can damage the central nervous system (CNS) and peripheral nervous system (PNS), leading to irreversible changes and causing functional deficits. Unlike in the CNS, damaged axons in peripheral nerves can be induced to regenerate in response to intrinsic cues and this process is triggered by a variety of inducers [1][2]. Proper axonal outgrowth in the adult CNS would be crucial for successful nerve regeneration and brain repair; however, an equilibrium between neuronal extrinsic and intrinsic factors is necessary to fully restore the injured neurons [3][4]. There are many identified mechanisms activated during neuronal injury, but due to complexity of the events regulating the subsequent regeneration, which involve multiple intrinsic signaling pathways, all the processes underlying the regeneration process have not been identified yet [5][6]. A wide variety of molecules are required in axonal regeneration, but because of the heterogeneity and functional diversity of neurons, they can also differentially affect the reactivation of specific tracts [7]. The efficiency of the regeneration process depends on the intrinsic properties of axonal outgrowth, the rate of the neuronal synthesis of proteins, cytoskeletal organization, axonal transport along the microtubules; all are critical for the regenerative response [3][8][9]. Of interest is the activation of numerous regeneration-associated genes (RAGs) after injury, and the participation of several transcription factors that are associated with regeneration [10][11][12].
Calcium ions, which physiologically play a very important function as second messengers, are also the inducers of cell death. It is well documented that the toxicity generated by increased, uncontrolled calcium levels in the cytosol after injury can lead to cell damage [13][14][15][16]. A significant component of the damage response process is a local translation in axons, which is essential for the regenerative effect. It enables the production of axonal regrowth molecules and induces regenerative pathways. For example, a moderate increase in Ca2+ is a factor that can stabilize the pool of F-actin required for the structural remodeling of spines. This multistep process involves the participation of several calcium-regulated proteins, including Ca2+/CaM kinase II and caldendrin [17][18][19]. Interestingly, calcium ions are also engaged in the restoration of neuronal homeostasis, which relies on calcium-regulated proteins expressed in the nervous system. Their specific role is determined by interactions with other functional proteins, which encompass Ca2+-regulated enzymes; proteins acting as Ca2+ sensors, represented by ubiquitous calmodulin (CaM) and CaM-like protein family; and Ca2+ buffers, which consist of many small, cytosolic proteins (i.e., parvalbumins, calbindins, calretinin) (for rev. [20][21]). Calmodulin appears to be the most recognized calcium sensor, and it can regulate over three hundred different targets with varying specific structural affinities [22][23][24][25] links Ca2+ signals to cellular functions through various effector proteins, including Ca2+/CaM-dependent kinases and phosphatases, or plasma membrane Ca2+-ATPase (PMCA) [26][27][28]. CaM binding by GAP-43 may also directly and indirectly affect regeneration process [29][30].
2. CaMKII
Calcium/calmodulin-dependent protein kinase II (CaMKII) is a member of the serine/threonine kinases family, and involved in cellular calcium signaling throughout the body. CaMKII regulates a vast array of functions within the brain, heart, epithelia, and immune system [31][32][33][34]. In the brain, where was first discovered, CaMKII plays a crucial role in neurotransmission, synaptic plasticity, memory, and learning [35]. As a one of the amplest proteins in neurons, is expressed in four isoforms: α and β are identified mainly there, δ in the cardiovascular system, and γ ubiquitously, encoded by four diverse genes [31][35]. CaMKII is a holoenzyme consisting of 12–14 subunits, forming a hetero- or homomeric structure, determining its unique properties [31].
Calcium influx initiates the binding of Ca2+ to CaM and as a complex bind to the CaMKII regulatory domain to induce a structural change, releasing the catalytic domain from the autoinhibitory region and exposing the substrate-binding site [36]. Ca2+/CaM binding results in autophosphorylation at T286 in CaMKIIα (Thr287 in CaMKIIβ, γ and ∂) in the autoinhibitory region, which enables phosphorylation of the neighboring activated subunit, causing Ca2+-independent CaMKII activity [31][36]. Interestingly, some CaMKIIγ subtypes, due to the nuclear localization sequence (NLS) signal, take part in shuttling the Ca2+/CaM complex to the nucleus to activate the cAMP-response element binding (CREB) protein in the brain [31].CaMKII, especially isoforms α and β, is a central coordinator of Ca2+ signal transduction in neurons and contributes to many physiological processes, like neurotransmitters metabolism, synaptic organization, long-term potentiation (LTP), and pathological processes, e.g., impaired learning, epilepsy, Alzheimer’s disease, ischemic stroke, and Parkinson’s disease [37]. The inability of axonal growth to occur in damaged neurons in the CNS is the main obstacle in the treatment of the diseases mentioned above [35]. It has been shown that calcium influx occurs immediately after axonal injury and promotes neuronal regeneration [38][39]. It is not surprising that CaMKII’s role in axonal regeneration has been proved in numerous studies.
The first reports demonstrated the promotion of neurite outgrowth in neuroblastoma lines when CaMKII was overexpressed or stimulated [40][41]. Likewise, pharmacological or genetic silencing inhibits CaMKII activity, leading to weakened axonal elongation in in vitro and in vivo studies or inducing the apoptosis and neuronal death of primary hippocampal neurons [35][42]. Moreover, activation of the kinase was relevant to the extension of prime neurons in vitro in embryonic CNS and mature PNS [35]. Axon regeneration, even of long-distance neurites, depends on the modulation of the cytoskeletal assembly at the nerve growth cone [43][44]. Since CaMKII colocalizes and operates with F-actin, a key cytoskeletal element, kinase activity may influence growth cone motility [45]. The inhibition of CaMKII destabilizes F-actin’s structure, and, in hippocampal neurons, overexpression leads to dendritic arborization, while silencing has the opposite effect [46][47].
Retinal ganglion cells (RGCs) are one of the most sensitive cells to changes in Ca2+ homeostasis during retinal ischemia or excitotoxicity. The first studies on CaMKII’s role in RGC survival and regeneration showed inconsistent results—chemically diminished CaMKII activity, under certain conditions, protects RGC, while in others it causes apoptosis and cellular hyperexcitability [48][49][50].
CaMKII has been implicated in several mechanisms that contribute to neuronal survival and recovery after ischemia. Previous reports have demonstrated evidence supporting the role of CaMKII activity in ischemic cell death, indicating that substantial neuroprotection can be achieved by inhibiting CaMKII. However, the broad inhibition of this pivotal kinase is challenging due to its involvement in various physiological processes like synaptic plasticity [51]. It was detected that the constitutive knockout of CaMKIIα provided protection against neuronal cell death following global cerebral ischemia in mice.
3. GAP-43
Growth associated protein 43 (GAP-43) is an exclusively neuronal protein connected to nerve development and regeneration, synaptic plasticity, axonal pathfinding, and neurotransmission [30]. The expression of GAP-43 is highly elevated in neuronal growth cones during synaptogenesis, and after the completion of the process, it declines in most brain areas, except in regions involved in learning and memory, like the neocortex and hippocampus in the adult brain [52][53][54]. In neurons, GAP-43 prevails in axon terminals, enabling actin cytoskeleton modulation [55][56]. The importance of GAP-43 in neurodevelopment was verified by homozygous knockout mice, which died early in their neonatal period due to axonal misguiding and defects in cortical topography [52][55][56][57][58]. GAP-43 seems to be crucial not only for the axons of differentiating neurons but also for the centrosome, and a lack of GAP-43 expression led to the mislocalization of centrosome and mitotic spindles during brain growth, causing a significant reduction in the cerebellum [59].
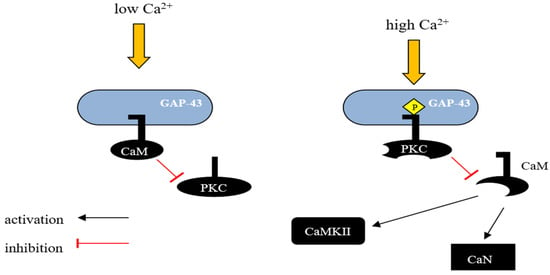
GAP-43 binds CaM more strongly at lower Ca2+ concentrations and releases it with a Ca2+ influx. Consequently, GAP-43 is considered as a temporary storage form place for CaM, which is was released at higher Ca2+ concentrations, sensitizing the downstream response (Figure 1) [29][60].
Figure 1. Schematic representation of GAP-43 activity regulation by calmodulin. GAP-43 can interact with CaM and the nature of this interaction depends on the intracellular concentration of Ca2+. In unstimulated neurons, where GAP-43 is present in large amounts, most of the CaM exists in a calcium-unbound form and is presumably located in the membrane fraction. In vivo, unphosphorylated GAP-43 binds to CaM at low Ca2+ concentrations in the cell, and this interaction inhibits the phosphorylation of Ser-41 by PKC, potentially protecting GAP-43 from activation in response to transient changes in the concentration of secondary messengers such as Ca2+, diacylglycerol, or arachidonic acid. However, when PKC is sufficiently activated, GAP-43 undergoes phosphorylation, leading to the release of CaM and blocking the possibility of its reassociation. This mechanism allows GAP-43 to remain in an active state even when the level of secondary messengers in the cell decreases. CaM—calmodulin, CaN—Ca2+ and calmodulin-dependent serine/threonine protein phosphatase 2B (Calcineurin), CaMKII—Ca2+/Calmodulin-dependent protein kinase II.
GAP-43 plays a crucial role in guiding the axons of retinal ganglion cells as they navigate from the optic chiasm to the optic tract [61]. The optic chiasm is known as a critical decision point for retinal axons [62]. Although GAP-43 is not essential for axonal outgrowth, it is crucial for the proper pathfinding of growth cones [62]. During the formation of the optic tract, GAP-43 facilitates the interaction of retinal axons with the lateral diencephalon [61].
GAP-43 plays an important role in nerve regeneration following injury and GAP-43 mRNA overexpression is closely associated with an improved regenerative capacity [63]. Elevated GAP-43 expression is also linked to axonal sprouting in the barrel cortex after a stroke [64]. Additionally, higher GAP-43 levels are connected to the optogenetic-induced functional recovery of the primary motor cortex after stroke [65].
4. Calcium-Binding Proteins
Ca2+ serves as a critical second messenger in neuronal signaling and can exert both positive and negative effects on neurite regeneration, involving a variety of proteins that transduce Ca2+ signals for neurite growth. Several identified Ca2+-binding proteins play a crucial role in controlling Ca2+ concentrations and may be essential for locally manipulating cellular calcium to facilitate neuronal repair. However, their neuron-specific expression and processes triggered following traumatic damage can lead to variations in their neuroregenerative properties and neurite growth dynamics. Within the category of calcium-binding proteins, there is a subset of EF-hand proteins related to calmodulin (CaM), which are highly expressed in the neuronal cell types of the brain. The EF-hand (helix-loop-helix motif) is one of the most frequently observed domains, and over 1000 have been identified based on their unique sequence signatures [66].
4.1. Oncomodulin
Oncomodulin (OCM) is a small Ca2+-binding protein (CaBP) belonging to the parvalbumins family, which are classical, small, mostly cytosolic and EF-hand-containing motifs molecules [67]. The EF-hand motif is characterized by an α-helix-loop-α-helix arrangement spanning approximately 30 amino acids, responsible for Ca2+ binding and triggering a conformational change leading to the activation or inactivation of target proteins [68]. OCM, as a member of the parvalbumin family, displays distinct characteristics in its ability to bind metal ions and sense calcium, resulting in functional differences when compared to other family members. Of the three domains in oncomodulin, EF1 cannot bind calcium ions due to the absence of key residues, but plays a vital role in OCM’s structural stability, EF2can bind a single Ca2+ and is characterized by a higher specificity for calcium ions, while EF3 accommodates both Ca2+and Mg2+. Upon exposure to calcium ions, OCM undergoes distinctive structural changes not observed in other parvalbumins. OCM features a cation binding site that accommodates both calcium and magnesium, with one being more Ca2+-specific. It is proposed that OCM might function as a calcium sensor or modulator under specific physiological conditions, rather than solely acting as a Ca2+ buffer [68].
OCM’s function appears ambiguous and cell-type-dependent when distinguishing it from other EF-hand CaBPs. Mammalian OCM expression is unique and confined to specific inner ear hair cells and specific immune cells [69][70][71]. In adult mammals, OCM can act as a Ca2+ buffer and likely influence outer hair cell (OHC) motility mechanisms, contributing to sensory hair cell function and playing a crucial role in maintaining hearing function and health [72][73].
4.2. Caldendrin
Among the interesting Ca2+-binding proteins is CaBP1, which, due to alternative splicing, forms three variants: caldendrin, CaBP1-S, and CaBP1-L [74][75]. Caldendrin has been shown to be highly expressed in different neuronal cell types, including the brain, retina, and inner ear [76][77]. Specifically, it is present in the synapses of cerebral cortical neurons, the cerebellum, hippocampus, and thalamus, as well as in the postsynaptic density of spine synapses [78][79]. Caldendrin has a unique bipartite structure with high homology to calmodulin, a ubiquitous EF-hand calcium sensor protein. However, in contrast to calmodulin, other Ca2+ sensor proteins have more specialized functions. Caldendrin possesses only two functional domains (EF3 and EF4) and two atypical domains (EF1 and EF2) [80]. The EF1 hand can bind Mg2+ with high affinity, whereas EF2 is non-functional. Two shorter isoforms of caldendrin detected in rats and humans differ in their sequences and are N-terminally myristoylated [75][79][81]. These proteins are mainly localized in the cytosol but are also present in the Golgi structures [82]. It should be underlined that caldendrin exhibits a high degree of conservation in humans, rats, and mice [83].
Caldendrin has been suggested to be involved in the processing of synaptic Ca2+ signaling [84]. A long-lasting increase in intracellular Ca2+ levels, either due to an influx through Ca2+ channels or a release from Ca2+ stores, is neurotoxic. However, some channels can be rapidly inactivated by the Ca2+-dependent inactivation process (CDI), which is a negative feedback mechanism important for regulating Ca2+ entry under both physiological and pathological conditions [85][86][87]. Physiologically, caldendrin increases the Ca2+influx throughL-type voltage-gated Ca2+ channels—Cav1.2 and Cav1.3—by interacting with their C-terminal domain [88][89][90][91][92]. In contrast, CaBP1 enhances the inactivation of Cav2.1 channels (P/Q-type) by interacting with the CaM-binding domain of the channel [93].
Caldendrin can also support the cytoskeleton network by stabilizing F-actin within the network’s spines through its interaction with cortactin, a protein implicated in actin filament nucleation and branching. This regulation contributes to spine stability and participates in the development of long-term memory and its storage [84]. Interestingly, cortactin activation appears to be caldendrin-specific and CaM-independent. The dynamics of spine morphology are attributed to the actin cytoskeleton that is highly concentrated in spines. However, the transport of organelles and their anchoring at dendritic spines also depend on F-actin-linked myosins [94].
A number of studies using caldendrin knockout animals have shown a more intensive regenerative growth of neurites, confirming the inhibitory action of this protein in the regeneration and elongation of neurites [95][96]. It is suggested that caldendrin may influence an initial phase of regeneration by altering the transcription of genes that promote axon outgrowth [96][97].
A crucial role in coordinating neuronal processes involves the formation of protein complexes named signalosomes, which are formed by scaffold proteins at specific subcellular locations. A notable example is a family of A-kinase anchoring proteins (AKAPs) that bind to protein kinase A (PKA) and other secondary messenger-regulated enzymes [98]. One of the essential elements for signal transduction is the protein AKAP79/150, which links receptors, channels, and other signaling proteins to physiological substrates and has been associated with the regulation of neurite growth [99]. At postsynaptic sites in neurons, AKAP79/150 can form complexes with protein kinases A and C, calcineurin, calmodulin, phosphatidylinositol 4,5-bisphosphate, and also with caldendrin [100].
One of the signaling pathways in neurons involves interactions between initial activity-dependent molecular changes at the synapse and the subsequent regulation of gene transcription in the nucleus. An interesting example of such communication is the contribution of Jacob, a protein messenger abundantly expressed in the brain, whichconnects NMDA-receptor-derived signalosomes to the transcription factor CREB [101][102][103]. The activation of synaptic NMDARs induces the expression of pro-survival genes, but the activation of extrasynaptic NMDARs initiates the expression of cell death genes [104]. The overexpression of Jacob triggers the expression of genes that induce neurodegeneration, whereas the nuclear knockdown of Jacob increases the phosphorylation of CREB and protected neurons from an extrasynaptic NMDA receptor-induced loss of synaptic contacts and neuronal cell death [105]. Caldendrin was shown to bind to Jacob’s nuclear localization signal in a Ca2+-dependent manner, and this interaction enabled the proper formation of the signalosome, representing a powerful regulatory mechanism of synapse-to-nucleus communication. Ultimately, significant changes in the morphology of the dendritic tree were generated [105].
4.3. Calneurons
Within the CaBP family, there also exists an interesting calciumsensor subfamily of proteins consisting of calneuron 1 (CaBP8) and calneuron 2 (CaBP7), which are abundant in the brain and exhibit developmental changes in expression [77][83][106]. They have 64% similarity and are present at high levels in distinct regions of the adult mammalian brain [107][108].
It is noteworthy that calneurons are highly conserved among different species, exhibiting 100% identity at the amino acid level between mice, rats, monkeys, and humans. This underscores their crucial cellular function. Both calneurons are distributed across various subcellular fractions, primarily localized in Golgi membranes, cytoplasm, and vesicles. In the mouse brain, calneuron 1 was found in significant amounts in the cerebellum, hippocampus, and cortex [109].
Additionally, calneurons exhibit sequence homology with calmodulin. In contrast to other calcium-binding proteins (CaBPs), calneurons display a distinct pattern of EF-hand inactivation, featuring active EF-hands 1 and 2 and inactive EF-hands 3 and 4 [106].
Less is known about functions of calneuron-1 and calneuron-2 [80]. As of publication, they have been found to regulate the activity of phosphatidylinositol 4-OH kinase IIIβ (PI4KIIIβ), which catalyzes the local synthesis of the phosphoinositides necessary for vesicle assembly in the trans-Golgi network (TGN) [110]. The activity of PI4KIIIβ at the Golgi membrane is a crucial initial step in trans-Golgi network-to-plasma-membrane trafficking. A primary and central regulator of PI4KIIIβ activity is NCS-1, a neuronal calcium sensor protein responsible for the rapid transduction of Ca2+ signals. NCS-1 is involved in numerous physiological neuronal functions, including exocytosis, the regulation of calcium channels, nuclear Ca2+ regulation, neurite outgrowth, and neuroprotection, as well as axonal regeneration in response to neuronal damage [111][112][113]. Therefore, it was interesting to find that both calneurons can directly associate with PI4KIIIβ both in vitro and in vivo in a Ca2+-independent manner [108].
4.4. Neuronal Calcium Sensor-1
Neuronal calcium sensor-1 (NCS-1) is a member of the NCS superfamily, characterized by four EF-hand domains. As with other members, the first EF-hand is unable to bind Ca2+, EF-2 serves as a Mg2+/Ca2+-binding domain, and the C-terminal domain contains Ca2+-binding EF-3 and EF-4 [114]. NCS-1 features an N-terminal myristoylation site facilitating its binding to cell membranes [115]. Ca2+ binding to NCS1 causes a structural rearrangement, modulating its affinity for target molecules [116]. In the brain, NCS-1 is widely expressed, with the highest abundance observed in neuronal tissues, particularly in the cortex, as well as in the hippocampus and dorsal root ganglion cells [117][118]. The specific expression patterns of NCS-1 in various brain regions, cell types, and subcellular areas can influence the availability of its unique target proteins.
To date, NCS-1 has been demonstrated to regulate various cellular functions, encompassing exocytosis, neurite outgrowth, neuroprotection, and axonal regeneration [119][120]. The functional diversity of NCS-1 arises from its interaction with numerous downstream targets. These include the binding to and regulation of the CaV2.1 of VGCCs, the enhancement of inositol 1,4,5-trisphosphate receptor activity, and the spatial and temporal control of phosphatidylinositol 4-phosphate levels through the activation of phosphatidylinositol 4-kinase III-β (PI-4Kβ). Additionally, NCS-1 contributes to the desensitization of dopamine type-2 receptors, among other pathways [121][122][123][124][125][126][127][128][129]. An intriguing relationship has been proposed regarding a molecular switch in the Ca2+ regulation of PI-4Kβ activity by calneurons and NCS-1 in Golgi membranes. Calneurons at low Ca2+ levels suppress PI-4Kβ activity and dominate in the regulation of this enzyme. As Ca2+ increases, the formation of the NCS-1/PI-4Kβ complex is promoted, potentially allowing for the override of the inhibition of PI-4Kβ activity imposed by calneurons [108].
5. Conclusions
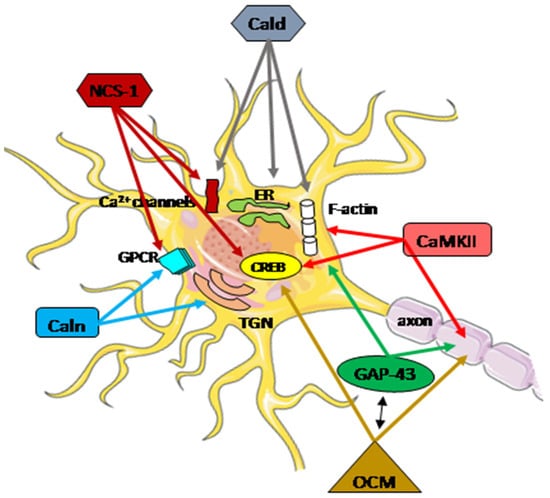
The highly diverse nature of the Ca2+ signaling in the nerve system is well established. At every stage of life, the functional adaptation of the molecular pathways in individual neurons is required to properly integrate external and internal factors. Disturbances in calcium homeostasis, resulting from a dysregulation of the ability to handle, store, and transfer information, contribute to the neuropathological processes in neurons, particularly after brain injury. The coordinated actions of all systems responsible for the maintenance and restoration of calcium homeostasis involve a complex interplay between functional proteins, including oncomodulin, caldendrin, calneuron, NCS-1, GAP-43, and CaM kinase II (Figure 2).
Figure 2. Schematic presentation of the regulatory function of Ca2+-associated proteins in neuron repair. A subtle regulatory control of Ca2+ signals plays a crucial role in integrating cellular mechanisms, and the cooperation of all potentially interacting partners is essential for specific neuronal functions. These functions encompass neurotransmitter release, proper neuronal transmission, activity-dependent gene transcription, endoplasmic reticulum (ER) targeting, Golgi-to-plasma-membrane trafficking, axonal growth, and cytokinesis. Arrows within the figure indicate potential direct sites and/or processes where specific Ca2+-associated proteins may be engaged. The activation or inhibition of neuron repair depends on the presence of partner proteins and the Ca2+ level. CaMKII actively participates in axon regeneration, colocalizes, and interacts with F-actin, activating the cAMP-response element-binding (CREB) protein. Phosphorylated GAP-43 stimulates F-actin polymerization and stability, leading to F-actin accumulation. GAP-43 also plays a pivotal role in axon outgrowth, neurotransmission, and synaptic plasticity. Oncomodulin (OCM) facilitates axon regeneration by operating within cellular signaling pathways, notably the CaMKII pathway, and is linked to the phosphorylation and activation of CREB. OCM contributes to the upregulation of selected RAGs, including Gap-43. Caldendrin (Cald) and two shorter CaBP1 forms regulate the Ca2+ influx through Ca2+ channels, including L-type and P/Q-type VGCC, and also TRPC5, which is implicated in neurite extension and growth cone morphology. Furthermore, Caldendrin, in a Ca2+-independent manner, could inhibit IP3 binding to inositol 1,4,5 trisphosphate receptors (IP3Rs), thereby blocking Ca2+ release from the ER. It also supports the cytoskeleton network by stabilizing F-actin within the spines.Calneuron-1 and Calneuron-2 (Caln) regulate the activity of phosphatidylinositol 4-OH kinase IIIβ, catalyzing the local synthesis of phosphoinositides necessary for vesicle assembly in the trans-Golgi network (TGN). Calneuron-1 can modulate G-protein-coupled receptor (GPCR) heteromers, such as adenosine A2A receptor (A2AR)-dopamine D2 receptor (D2R) and, as recently shown, cannabinoid CB1 receptor. Neuronal calcium sensor-1 (NCS-1) binds to and regulates the CaV2.1 of VGCCs and TRPC5, enhancing the activity of IP3Rs and PI-4Kβ in Golgi membranes. Additionally, it contributes to the desensitization of D2Rs. The NCS-1-induced activation of cytosolic CaMKII-α can regulate CREB.
References
- Tedeschi, A.; Bradke, F. Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr. Opin. Neurobiol. 2017, 42, 118–127.
- Shamoun, F.; Shamoun, V.; Akhavan, A.; Tuffaha, S.H. Target Receptors of Regenerating Nerves: Neuroma Formation and Current Treatment Options. Front. Mol. Neurosci. 2022, 15, 859221.
- Bradke, F. Mechanisms of Axon Growth and Regeneration: Moving between Development and Disease. J. Neurosci. 2022, 42, 8393–8405.
- Dubový, P.; Klusáková, I.; Hradilová-Svíženská, I.; Joukal, M. Expression of Regeneration-Associated Proteins in Primary Sensory Neurons and Regenerating Axons After Nerve Injury—An Overview. Anat. Rec. 2018, 301, 1618–1627.
- Makwana, M.; Raivich, G. Molecular mechanisms in successful peripheral regeneration. FEBS J. 2005, 272, 2628–2638.
- Raivich, G.; Makwana, M. The making of successful axonal regeneration: Genes, molecules and signal transduction pathways. Brain Res. Rev. 2007, 53, 287–311.
- Woodworth, M.B.; Greig, L.C.; Goldberg, J.L. Intrinsic and Induced Neuronal Regeneration in the Mammalian Retina. Antioxid. Redox Signal. 2023, 39, 1039–1052.
- Di Giaimo, R.; Penna, E.; Pizzella, A.; Cirillo, R.; Perrone-Capano, C.; Crispino, M. Cross Talk at the Cytoskeleton-Plasma Membrane Interface: Impact on Neuronal Morphology and Functions. Int. J. Mol. Sci. 2020, 21, 9133.
- Blanquie, O.; Bradke, F. Cytoskeleton dynamics in axon regeneration. Curr. Opin. Neurobiol. 2018, 51, 60–69.
- An, J.; Chen, B.; Tian, D.; Guo, Y.; Yan, Y.; Yang, H. Regulation of Neurogenesis and Neuronal Differentiation by Natural Compounds. Curr. Stem Cell Res. Ther. 2022, 17, 756–771.
- Patodia, S.; Raivich, G. Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 2012, 5, 8.
- Zhang, Y.; Zhao, Q.; Chen, Q.; Xu, L.; Yi, S. Transcriptional Control of Peripheral Nerve Regeneration. Mol. Neurobiol. 2023, 60, 329–341.
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590.
- Nedergaard, M.; Verkhratsky, A. Calcium dyshomeostasis and pathological calcium signalling in neurological diseases. Cell Calcium 2010, 47, 101–102.
- Uryash, A.; Flores, V.; Adams, J.A.; Allen, P.D.; Lopez, J.R. Memory and Learning Deficits Are Associated With Ca. Front. Aging Neurosci. 2020, 12, 224.
- Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33.
- Khan, S.; Conte, I.; Carter, T.; Bayer, K.U.; Molloy, J.E. Multiple CaMKII Binding Modes to the Actin Cytoskeleton Revealed by Single-Molecule Imaging. Biophys. J. 2016, 111, 395–408.
- Kim, K.; Lakhanpal, G.; Lu, H.E.; Khan, M.; Suzuki, A.; Hayashi, M.K.; Narayanan, R.; Luyben, T.T.; Matsuda, T.; Nagai, T.; et al. A Temporary Gating of Actin Remodeling during Synaptic Plasticity Consists of the Interplay between the Kinase and Structural Functions of CaMKII. Neuron 2015, 87, 813–826.
- Wang, Q.; Chen, M.; Schafer, N.P.; Bueno, C.; Song, S.S.; Hudmon, A.; Wolynes, P.G.; Waxham, M.N.; Cheung, M.S. Assemblies of calcium/calmodulin-dependent kinase II with actin and their dynamic regulation by calmodulin in dendritic spines. Proc. Natl. Acad. Sci. USA 2019, 116, 18937–18942.
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051.
- Eisner, D.; Neher, E.; Taschenberger, H.; Smith, G. Physiology of intracellular calcium buffering. Physiol. Rev. 2023, 103, 2767–2845.
- Liu, F.; Chu, X.; Lu, H.P.; Wang, J. Molecular mechanism of multispecific recognition of Calmodulin through conformational changes. Proc. Natl. Acad. Sci. USA 2017, 114, E3927–E3934.
- Kursula, P. The many structural faces of calmodulin: A multitasking molecular jackknife. Amino Acids 2014, 46, 2295–2304.
- Shukla, D.; Peck, A.; Pande, V.S. Conformational heterogeneity of the calmodulin binding interface. Nat. Commun. 2016, 7, 10910.
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 2018, 14, e1006072.
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193.
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565.
- Mantilla, G.; Peréz-Gordones, M.C.; Cisneros-Montufar, S.; Benaim, G.; Navarro, J.C.; Mendoza, M.; Ramírez-Iglesias, J.R. Structural Analysis and Diversity of Calmodulin-Binding Domains in Membrane and Intracellular Ca. J. Membr. Biol. 2023, 256, 159–174.
- Denny, J.B. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304.
- Chung, D.; Shum, A.; Caraveo, G. GAP-43 and BASP1 in Axon Regeneration: Implications for the Treatment of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 567537.
- Zhang, X.; Connelly, J.; Levitan, E.S.; Sun, D.; Wang, J.Q. Calcium/Calmodulin-Dependent Protein Kinase II in Cerebrovascular Diseases. Transl. Stroke Res. 2021, 12, 513–529.
- Anderson, M.E. Calmodulin kinase signaling in heart: An intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 2005, 106, 39–55.
- Lin, M.Y.; Zal, T.; Ch’en, I.L.; Gascoigne, N.R.; Hedrick, S.M. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: From activation to unresponsiveness. J. Immunol. 2005, 174, 5583–5592.
- McGargill, M.A.; Sharp, L.L.; Bui, J.D.; Hedrick, S.M.; Calbo, S. Active Ca2+/calmodulin-dependent protein kinase II gamma B impairs positive selection of T cells by modulating TCR signaling. J. Immunol. 2005, 175, 656–664.
- Xi, F.; Xu, R.J.; Xu, J.H.; Ma, J.J.; Wang, W.H.; Wang, F.; Ma, Y.X.; Qi, S.B.; Zhang, H.C.; Zhang, H.N.; et al. Calcium/calmodulin-dependent protein kinase II regulates mammalian axon growth by affecting F-actin length in growth cone. J. Cell. Physiol. 2019, 234, 23053–23065.
- Nicole, O.; Pacary, E. CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases. Int. J. Mol. Sci. 2020, 21, 7272.
- Rostas, J.A.P.; Skelding, K.A. Calcium/Calmodulin-Stimulated Protein Kinase II (CaMKII): Different Functional Outcomes from Activation, Depending on the Cellular Microenvironment. Cells 2023, 12, 401.
- Mar, F.M.; Bonni, A.; Sousa, M.M. Cell intrinsic control of axon regeneration. EMBO Rep. 2014, 15, 254–263.
- Ghosh-Roy, A.; Wu, Z.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 2010, 30, 3175–3183.
- Goshima, Y.; Ohsako, S.; Yamauchi, T. Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. J. Neurosci. 1993, 13, 559–567.
- Sogawa, Y.; Yoshimura, Y.; Otaka, A.; Yamauchi, T. Ca2+-independent activity of Ca2+/calmodulin-dependent protein kinase II involved in stimulation of neurite outgrowth in neuroblastoma cells. Brain Res. 2000, 881, 165–175.
- Wang, J.; Xu, X.; Jia, W.; Zhao, D.; Boczek, T.; Gao, Q.; Wang, Q.; Fu, Y.; He, M.; Shi, R.; et al. Calcium-/Calmodulin-Dependent Protein Kinase II (CaMKII) Inhibition Induces Learning and Memory Impairment and Apoptosis. Oxid. Med. Cell. Longev. 2021, 2021, 4635054.
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931.
- Hur, E.M.; Yang, I.H.; Kim, D.H.; Byun, J.; Saijilafu; Xu, W.L.; Nicovich, P.R.; Cheong, R.; Levchenko, A.; Thakor, N.; et al. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 5057–5062.
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 1998, 21, 593–606.
- Easley, C.A.; Faison, M.O.; Kirsch, T.L.; Lee, J.A.; Seward, M.E.; Tombes, R.M. Laminin activates CaMK-II to stabilize nascent embryonic axons. Brain Res. 2006, 1092, 59–68.
- Fink, C.C.; Bayer, K.U.; Myers, J.W.; Ferrell, J.E.; Schulman, H.; Meyer, T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 2003, 39, 283–297.
- Goebel, D.J. Selective blockade of CaMKII-alpha inhibits NMDA-induced caspase-3-dependent cell death but does not arrest PARP-1 activation or loss of plasma membrane selectivity in rat retinal neurons. Brain Res. 2009, 1256, 190–204.
- Laabich, A.; Li, G.; Cooper, N.G. Calcium/calmodulin-dependent protein kinase II containing a nuclear localizing signal is altered in retinal neurons exposed to N-methyl-D-aspartate. Brain Res. Mol. Brain Res. 2000, 76, 253–265.
- Ashpole, N.M.; Song, W.; Brustovetsky, T.; Engleman, E.A.; Brustovetsky, N.; Cummins, T.R.; Hudmon, A. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem. 2012, 287, 8495–8506.
- Griem-Krey, N.; Clarkson, A.N.; Wellendorph, P. CaMKIIα as a Promising Drug Target for Ischemic Grey Matter. Brain Sci. 2022, 12, 1639.
- Frey, D.; Laux, T.; Xu, L.; Schneider, C.; Caroni, P. Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J. Cell Biol. 2000, 149, 1443–1454.
- McGuire, C.B.; Snipes, G.J.; Norden, J.J. Light-microscopic immunolocalization of the growth- and plasticity-associated protein GAP-43 in the developing rat brain. Brain Res. 1988, 469, 277–291.
- Benowitz, L.I.; Apostolides, P.J.; Perrone-Bizzozero, N.; Finklestein, S.P.; Zwiers, H. Anatomical distribution of the growth-associated protein GAP-43/B-50 in the adult rat brain. J. Neurosci. 1988, 8, 339–352.
- Widmer, F.; Caroni, P. Identification, localization, and primary structure of CAP-23, a particle-bound cytosolic protein of early development. J. Cell Biol. 1990, 111, 3035–3047.
- Meiri, K.F.; Pfenninger, K.H.; Willard, M.B. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc. Natl. Acad. Sci. USA 1986, 83, 3537–3541.
- Shen, Y.; Mani, S.; Donovan, S.L.; Schwob, J.E.; Meiri, K.F. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J. Neurosci. 2002, 22, 239–247.
- Metz, G.A.; Schwab, M.E. Behavioral characterization in a comprehensive mouse test battery reveals motor and sensory impairments in growth-associated protein-43 null mutant mice. Neuroscience 2004, 129, 563–574.
- Mishra, R.; Gupta, S.K.; Meiri, K.F.; Fong, M.; Thostrup, P.; Juncker, D.; Mani, S. GAP-43 is key to mitotic spindle control and centrosome-based polarization in neurons. Cell Cycle 2008, 7, 348–357.
- Mosevitsky, M.I. Nerve ending “signal” proteins GAP-43, MARCKS, and BASP1. Int. Rev. Cytol. 2005, 245, 245–325.
- Zhang, F.; Lu, C.; Severin, C.; Sretavan, D.W. GAP-43 mediates retinal axon interaction with lateral diencephalon cells during optic tract formation. Development 2000, 127, 969–980.
- Strittmatter, S.M.; Fankhauser, C.; Huang, P.L.; Mashimo, H.; Fishman, M.C. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell 1995, 80, 445–452.
- Skene, J.H.; Willard, M. Changes in axonally transported proteins during axon regeneration in toad retinal ganglion cells. J. Cell Biol. 1981, 89, 86–95.
- Carmichael, S.T.; Archibeque, I.; Luke, L.; Nolan, T.; Momiy, J.; Li, S. Growth-associated gene expression after stroke: Evidence for a growth-promoting region in peri-infarct cortex. Exp. Neurol. 2005, 193, 291–311.
- Cheng, M.Y.; Wang, E.H.; Woodson, W.J.; Wang, S.; Sun, G.; Lee, A.G.; Arac, A.; Fenno, L.E.; Deisseroth, K.; Steinberg, G.K. Optogenetic neuronal stimulation promotes functional recovery after stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 12913–12918.
- Henikoff, S. ENCODE and our very busy genome. Nat. Genet. 2007, 39, 817–818.
- Schwaller, B. Cytosolic Ca. Cold Spring Harb. Perspect. Biol. 2020, 12, a035543.
- Climer, L.K.; Cox, A.M.; Reynolds, T.J.; Simmons, D.D. Oncomodulin: The Enigmatic Parvalbumin Protein. Front. Mol. Neurosci. 2019, 12, 235.
- Yang, D.; Thalmann, I.; Thalmann, R.; Simmons, D.D. Expression of alpha and beta parvalbumin is differentially regulated in the rat organ of corti during development. J. Neurobiol. 2004, 58, 479–492.
- Yin, Y.; Cui, Q.; Gilbert, H.Y.; Yang, Y.; Yang, Z.; Berlinicke, C.; Li, Z.; Zaverucha-do-Valle, C.; He, H.; Petkova, V.; et al. Oncomodulin links inflammation to optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 19587–19592.
- Kurimoto, T.; Yin, Y.; Habboub, G.; Gilbert, H.Y.; Li, Y.; Nakao, S.; Hafezi-Moghadam, A.; Benowitz, L.I. Neutrophils express oncomodulin and promote optic nerve regeneration. J. Neurosci. 2013, 33, 14816–14824.
- Tong, B.; Hornak, A.J.; Maison, S.F.; Ohlemiller, K.K.; Liberman, M.C.; Simmons, D.D. Oncomodulin, an EF-Hand Ca2+ Buffer, Is Critical for Maintaining Cochlear Function in Mice. J. Neurosci. 2016, 36, 1631–1635.
- Hackney, C.M.; Mahendrasingam, S.; Penn, A.; Fettiplace, R. The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J. Neurosci. 2005, 25, 7867–7875.
- Seidenbecher, C.I.; Langnaese, K.; Sanmartí-Vila, L.; Boeckers, T.M.; Smalla, K.H.; Sabel, B.A.; Garner, C.C.; Gundelfinger, E.D.; Kreutz, M.R. Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J. Biol. Chem. 1998, 273, 21324–21331.
- Laube, G.; Seidenbecher, C.I.; Richter, K.; Dieterich, D.C.; Hoffmann, B.; Landwehr, M.; Smalla, K.H.; Winter, C.; Böckers, T.M.; Wolf, G.; et al. The neuron-specific Ca2+-binding protein caldendrin: Gene structure, splice isoforms, and expression in the rat central nervous system. Mol. Cell Neurosci. 2002, 19, 459–475.
- Seidenbecher, C.I.; Reissner, C.; Kreutz, M.R. Caldendrins in the inner retina. Adv. Exp. Med. Biol. 2002, 514, 451–463.
- Haeseleer, F.; Sokal, I.; Verlinde, C.L.; Erdjument-Bromage, H.; Tempst, P.; Pronin, A.N.; Benovic, J.L.; Fariss, R.N.; Palczewski, K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 2000, 275, 1247–1260.
- Bernstein, H.G.; Seidenbecher, C.I.; Smalla, K.H.; Gundelfinger, E.D.; Bogerts, B.; Kreutz, M.R. Distribution and cellular localization of caldendrin immunoreactivity in adult human forebrain. J. Histochem. Cytochem. 2003, 51, 1109–1112.
- Kim, K.Y.; Scholl, E.S.; Liu, X.; Shepherd, A.; Haeseleer, F.; Lee, A. Localization and expression of CaBP1/caldendrin in the mouse brain. Neuroscience 2014, 268, 33–47.
- Mundhenk, J.; Fusi, C.; Kreutz, M.R. Caldendrin and Calneurons-EF-Hand CaM-Like Calcium Sensors With Unique Features and Specialized Neuronal Functions. Front. Mol. Neurosci. 2019, 12, 16.
- Kiran, U.; Regur, P.; Kreutz, M.R.; Sharma, Y.; Chakraborty, A. Intermotif Communication Induces Hierarchical Ca. Biochemistry 2017, 56, 2467–2476.
- Haynes, L.P.; McCue, H.V.; Burgoyne, R.D. Evolution and functional diversity of the Calcium Binding Proteins (CaBPs). Front. Mol. Neurosci. 2012, 5, 9.
- McCue, H.V.; Haynes, L.P.; Burgoyne, R.D. Bioinformatic analysis of CaBP/calneuron proteins reveals a family of highly conserved vertebrate Ca2+-binding proteins. BMC Res. Notes 2010, 3, 118.
- Mikhaylova, M.; Bär, J.; van Bommel, B.; Schätzle, P.; YuanXiang, P.; Raman, R.; Hradsky, J.; Konietzny, A.; Loktionov, E.Y.; Reddy, P.P.; et al. Caldendrin Directly Couples Postsynaptic Calcium Signals to Actin Remodeling in Dendritic Spines. Neuron 2018, 97, 1110–1125.e14.
- Hall, D.D.; Dai, S.; Tseng, P.Y.; Malik, Z.; Nguyen, M.; Matt, L.; Schnizler, K.; Shephard, A.; Mohapatra, D.P.; Tsuruta, F.; et al. Competition between α-actinin and Ca2+-calmodulin controls surface retention of the L-type Ca2+ channel Ca(V)1.2. Neuron 2013, 78, 483–497.
- Oliveria, S.F.; Dittmer, P.J.; Youn, D.H.; Dell’Acqua, M.L.; Sather, W.A. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J. Neurosci. 2012, 32, 15328–15337.
- Christel, C.; Lee, A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 2012, 1820, 1243–1252.
- Zhou, H.; Yu, K.; McCoy, K.L.; Lee, A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J. Biol. Chem. 2005, 280, 29612–29619.
- Tippens, A.L.; Lee, A. Caldendrin, a neuron-specific modulator of Cav/1.2 (L-type) Ca2+ channels. J. Biol. Chem. 2007, 282, 8464–8473.
- Oz, S.; Benmocha, A.; Sasson, Y.; Sachyani, D.; Almagor, L.; Lee, A.; Hirsch, J.A.; Dascal, N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J. Biol. Chem. 2013, 288, 12680–12691.
- Ames, J.B. L-Type Ca. Biomolecules 2021, 11, 1811.
- Findeisen, F.; Minor, D.L. Structural basis for the differential effects of CaBP1 and calmodulin on Ca(V)1.2 calcium-dependent inactivation. Structure 2010, 18, 1617–1631.
- Lee, A.; Westenbroek, R.E.; Haeseleer, F.; Palczewski, K.; Scheuer, T.; Catterall, W.A. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 2002, 5, 210–217.
- Kapitein, L.C.; van Bergeijk, P.; Lipka, J.; Keijzer, N.; Wulf, P.S.; Katrukha, E.A.; Akhmanova, A.; Hoogenraad, C.C. Myosin-V opposes microtubule-based cargo transport and drives directional motility on cortical actin. Curr. Biol. 2013, 23, 828–834.
- Yang, T.; Choi, J.E.; Soh, D.; Tobin, K.; Joiner, M.L.; Hansen, M.; Lee, A. CaBP1 regulates Ca. Mol. Cell. Neurosci. 2018, 88, 342–352.
- Lopez, J.A.; Yamamoto, A.; Vecchi, J.T.; Hagen, J.; Lee, K.; Sonka, M.; Hansen, M.R.; Lee, A. Caldendrin represses neurite regeneration and growth in dorsal root ganglion neurons. Sci. Rep. 2023, 13, 2608.
- Wu, J.; Xie, X. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006, 7, R85.
- Wild, A.R.; Dell’Acqua, M.L. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther. 2018, 185, 99–121.
- Zhang, J.; Shapiro, M.S. Mechanisms and dynamics of AKAP79/150-orchestrated multi-protein signalling complexes in brain and peripheral nerve. J. Physiol. 2016, 594, 31–37.
- Seeger, C.; Gorny, X.; Reddy, P.P.; Seidenbecher, C.; Danielson, U.H. Kinetic and mechanistic differences in the interactions between caldendrin and calmodulin with AKAP79 suggest different roles in synaptic function. J. Mol. Recognit. 2012, 25, 495–503.
- Karpova, A.; Mikhaylova, M.; Bera, S.; Bär, J.; Reddy, P.P.; Behnisch, T.; Rankovic, V.; Spilker, C.; Bethge, P.; Sahin, J.; et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 2013, 152, 1119–1133.
- Mikhaylova, M.; Karpova, A.; Bär, J.; Bethge, P.; YuanXiang, P.; Chen, Y.; Zuschratter, W.; Behnisch, T.; Kreutz, M.R. Cellular distribution of the NMDA-receptor activated synapto-nuclear messenger Jacob in the rat brain. Brain Struct. Funct. 2014, 219, 843–860.
- Grochowska, K.M.; Bär, J.; Gomes, G.M.; Kreutz, M.R.; Karpova, A. Jacob, a Synapto-Nuclear Protein Messenger Linking N-methyl-D-aspartate Receptor Activation to Nuclear Gene Expression. Front. Synaptic Neurosci. 2021, 13, 787494.
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696.
- Dieterich, D.C.; Karpova, A.; Mikhaylova, M.; Zdobnova, I.; König, I.; Landwehr, M.; Kreutz, M.; Smalla, K.H.; Richter, K.; Landgraf, P.; et al. Caldendrin-Jacob: A protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 2008, 6, e34.
- Mikhaylova, M.; Sharma, Y.; Reissner, C.; Nagel, F.; Aravind, P.; Rajini, B.; Smalla, K.H.; Gundelfinger, E.D.; Kreutz, M.R. Neuronal Ca2+ signaling via caldendrin and calneurons. Biochim. Biophys. Acta 2006, 1763, 1229–1237.
- Hradsky, J.; Bernstein, H.G.; Marunde, M.; Mikhaylova, M.; Kreutz, M.R. Alternative splicing, expression and cellular localization of Calneuron-1 in the rat and human brain. J. Histochem. Cytochem. 2015, 63, 793–804.
- Mikhaylova, M.; Reddy, P.P.; Munsch, T.; Landgraf, P.; Suman, S.K.; Smalla, K.H.; Gundelfinger, E.D.; Sharma, Y.; Kreutz, M.R. Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc. Natl. Acad. Sci. USA 2009, 106, 9093–9098.
- Wu, Y.Q.; Lin, X.; Liu, C.M.; Jamrich, M.; Shaffer, L.G. Identification of a human brain-specific gene, calneuron 1, a new member of the calmodulin superfamily. Mol. Genet. Metab. 2001, 72, 343–350.
- Rajamanoharan, D.; McCue, H.V.; Burgoyne, R.D.; Haynes, L.P. Modulation of phosphatidylinositol 4-phosphate levels by CaBP7 controls cytokinesis in mammalian cells. Mol. Biol. Cell 2015, 26, 1428–1439.
- Haynes, L.P.; Tepikin, A.V.; Burgoyne, R.D. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J. Biol. Chem. 2004, 279, 547–555.
- de Barry, J.; Janoshazi, A.; Dupont, J.L.; Procksch, O.; Chasserot-Golaz, S.; Jeromin, A.; Vitale, N. Functional implication of neuronal calcium sensor-1 and phosphoinositol 4-kinase-beta interaction in regulated exocytosis of PC12 cells. J. Biol. Chem. 2006, 281, 18098–18111.
- Yip, P.K.; Wong, L.F.; Sears, T.A.; Yáñez-Muñoz, R.J.; McMahon, S.B. Cortical overexpression of neuronal calcium sensor-1 induces functional plasticity in spinal cord following unilateral pyramidal tract injury in rat. PLoS Biol. 2010, 8, e1000399.
- Bourne, Y.; Dannenberg, J.; Pollmann, V.; Marchot, P.; Pongs, O. Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1). J. Biol. Chem. 2001, 276, 11949–11955.
- McFerran, B.W.; Weiss, J.L.; Burgoyne, R.D. Neuronal Ca2+)sensor 1. Characterization of the myristoylated protein, its cellular effects in permeabilized adrenal chromaffin cells, Ca2+-independent membrane association, and interaction with binding proteins, suggesting a role in rapid Ca2+ signal transduction. J. Biol. Chem. 1999, 274, 30258–30265.
- Aravind, P.; Chandra, K.; Reddy, P.P.; Jeromin, A.; Chary, K.V.; Sharma, Y. Regulatory and structural EF-hand motifs of neuronal calcium sensor-1: Mg 2+ modulates Ca2+ binding, Ca2+-induced conformational changes, and equilibrium unfolding transitions. J. Mol. Biol. 2008, 376, 1100–1115.
- Chen, C.; Yu, L.; Zhang, P.; Jiang, J.; Zhang, Y.; Chen, X.; Wu, Q.; Zhao, S. Human neuronal calcium sensor-1 shows the highest expression level in cerebral cortex. Neurosci. Lett. 2002, 319, 67–70.
- Saab, B.J.; Georgiou, J.; Nath, A.; Lee, F.J.; Wang, M.; Michalon, A.; Liu, F.; Mansuy, I.M.; Roder, J.C. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 2009, 63, 643–656.
- Dason, J.S.; Romero-Pozuelo, J.; Atwood, H.L.; Ferrús, A. Multiple roles for frequenin/NCS-1 in synaptic function and development. Mol. Neurobiol. 2012, 45, 388–402.
- Weiss, J.L.; Hui, H.; Burgoyne, R.D. Neuronal calcium sensor-1 regulation of calcium channels, secretion, and neuronal outgrowth. Cell. Mol. Neurobiol. 2010, 30, 1283–1292.
- Lian, L.Y.; Pandalaneni, S.R.; Todd, P.A.; Martin, V.M.; Burgoyne, R.D.; Haynes, L.P. Demonstration of binding of neuronal calcium sensor-1 to the cav2.1 p/q-type calcium channel. Biochemistry 2014, 53, 6052–6062.
- Tsujimoto, T.; Jeromin, A.; Saitoh, N.; Roder, J.C.; Takahashi, T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 2002, 295, 2276–2279.
- Iketani, M.; Imaizumi, C.; Nakamura, F.; Jeromin, A.; Mikoshiba, K.; Goshima, Y.; Takei, K. Regulation of neurite outgrowth mediated by neuronal calcium sensor-1 and inositol 1,4,5-trisphosphate receptor in nerve growth cones. Neuroscience 2009, 161, 743–752.
- Schlecker, C.; Boehmerle, W.; Jeromin, A.; DeGray, B.; Varshney, A.; Sharma, Y.; Szigeti-Buck, K.; Ehrlich, B.E. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J. Clin. Investig. 2006, 116, 1668–1674.
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361.
- Nguyen, L.D.; Petri, E.T.; Huynh, L.K.; Ehrlich, B.E. Characterization of NCS1-InsP3R1 interaction and its functional significance. J. Biol. Chem. 2019, 294, 18923–18933.
- Haynes, L.P.; Thomas, G.M.; Burgoyne, R.D. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J. Biol. Chem. 2005, 280, 6047–6054.
- Kabbani, N.; Negyessy, L.; Lin, R.; Goldman-Rakic, P.; Levenson, R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 2002, 22, 8476–8486.
- Haynes, L.P.; Sherwood, M.W.; Dolman, N.J.; Burgoyne, R.D. Specificity, promiscuity and localization of ARF protein interactions with NCS-1 and phosphatidylinositol-4 kinase-III beta. Traffic 2007, 8, 1080–1092.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
368
Revisions:
2 times
(View History)
Update Date:
08 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No