Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tomoya Maeda | -- | 3501 | 2024-01-29 11:45:37 | | | |
| 2 | Camila Xu | Meta information modification | 3501 | 2024-01-30 01:11:03 | | | | |
| 3 | Camila Xu | + 4 word(s) | 3505 | 2024-02-05 02:51:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Maeda, T.; Furusawa, C. Insights into AMR Mechanisms from Laboratory Evolution. Encyclopedia. Available online: https://encyclopedia.pub/entry/54486 (accessed on 08 August 2026).
Maeda T, Furusawa C. Insights into AMR Mechanisms from Laboratory Evolution. Encyclopedia. Available at: https://encyclopedia.pub/entry/54486. Accessed August 08, 2026.
Maeda, Tomoya, Chikara Furusawa. "Insights into AMR Mechanisms from Laboratory Evolution" Encyclopedia, https://encyclopedia.pub/entry/54486 (accessed August 08, 2026).
Maeda, T., & Furusawa, C. (2024, January 29). Insights into AMR Mechanisms from Laboratory Evolution. In Encyclopedia. https://encyclopedia.pub/entry/54486
Maeda, Tomoya and Chikara Furusawa. "Insights into AMR Mechanisms from Laboratory Evolution." Encyclopedia. Web. 29 January, 2024.
Copy Citation
Laboratory evolution studies provide invaluable insights into the dynamic landscape of resistance, revealing intricate details about the evolutionary trajectories, population dynamics, and metabolic adaptations shaping bacterial responses to drug exposure. The complex interplay of cross-resistance and collateral sensitivity in the evolution of antimicrobial resistance (AMR) has been intensively analyzed using E. coli as a model bacterium.
antimicrobial resistance
laboratory evolution
collateral sensitivity
1. Genotype-Phenotype Relationships in Antimicrobial Resistance (AMR) Evolution
Lázár et al. conducted laboratory evolution of E. coli by exposing cells to increasing dosages of 12 antibiotics [1]. This study revealed convergent molecular evolution across antibiotics, mutations conferring resistance enhancing sensitivity to other drugs, and the identification of chemogenomic profile similarity as a predictor of cross-resistance [1]. Similar results were observed in other laboratory evolution studies [2][3]. Despite diverse initial conditions, similar mutations and resistance mechanisms emerged under antibiotic selection. Mutations that confer resistance to a specific antibiotic often simultaneously enhance sensitivity to many other drugs, suggesting that resistance evolution is driven by mutations with broad consequence [1][2][3]. Approximately 27% of the observed mutations resulted in proteins with compromised or no activities, highlighting the role of loss-of-function mutations in antibiotic adaptation [1]. Cross-resistance patterns are strongly influenced by chemogenomic profile similarity between antibiotics, indicating that exposure to a single antibiotic often leads to multidrug resistance. Interestingly, cross-resistance between two antibiotics is largely independent of whether they show synergistic effects in combination [1].
To explore the complex relationship between genotype and phenotype in antibiotic-resistant E. coli strains, Suzuki et al. also conducted laboratory evolution of E. coli’s resistance to 11 antibiotics using a serial transfer method by selecting a population showing the growth under the highest drug concentration possible [2]. Through 90-day laboratory evolution and integrated transcriptome–genome analyses, the study revealed that resistance acquisition to one drug significantly alters susceptibility to others. They demonstrated that a simple linear model can quantitatively predict the resistance of 25 drugs via the expression changes of only eight genes [2]. The eight genes were acrB, encoding a subunit of a multi-drug efflux pump; ompF, encoding an outer membrane porin protein; cyoC, encoding the subunit of cytochrome bo3 terminal oxidase; pps, encoding the phosphoenolpyruvate synthase; tsx, encoding a nucleoside channel, a receptor of phage T6 and colicin K; oppA, encoding an oligopeptide transporter; folA, encoding the dihydrofolate reductase; and pntB, encoding the beta subunit of pyridine nucleotide transhydrogenase [2].
To understand the constraints shaping the evolution of AMR, a systematic investigation of evolutionary constraints through high-throughput laboratory evolution of E. coli was conducted with the addition of 95 antibacterial chemicals covering a wide range of action mechanisms [3]. Machine learning techniques were employed to analyze phenotype–genotype data, revealing low-dimensional phenotypic states and trade-off relationships associated with drug resistance. The findings provide insights into the intricate interplay between genomic, transcriptomic, and resistance profiles, contributing to a comprehensive understanding of evolutionary constraints in adaptive evolution [3]. Since E. coli’s evolutionary dynamics were attributable to a relatively small number of intracellular states, this indicates that E. coli is equipped with only a limited number of strategies for antibiotic resistance [3]. This study also quantified collateral responses to other antibiotics and found that 336 and 157 pairs of drugs within the 2162 combinations exhibited cross-resistance and collateral sensitivity, respectively [3]. Heteroresistance, a prevalent phenotype in clinically isolated strains to date [4][5], was observed in 15 out of 33 pairs in the laboratory-evolved strains, suggesting its frequent occurrence in the process of acquiring resistance [3]. It was suggested that the perturbation of metabolic activity by metabolic inhibitors, reactive oxygen species generation, and alteration of cytoplasmic membrane permeabilization by cationic peptides could serve as possible strategies to suppress antibiotic resistance [3].
Laboratory evolution also uncovered the often-overlooked area of metabolic adaptations in response to antibiotic treatment, shedding light on how alterations in cellular metabolism can contribute to antibiotic resistance [6]. This study emphasized the importance of population-level analyses in understanding the evolutionary landscape in response to drug treatment. This study aimed to maximize metabolic adaptation rather than growth adaptation in laboratory evolution protocols [6]. This shift in dynamics allowed for a more comprehensive view of antibiotic-specific metabolic variants, revealing underappreciated genes related to central carbon and energy metabolism [6]. Mutations in metabolic genes that arose in response to antibiotic treatment were found in multiple independent populations and responses to different drugs [6]. Particularly, a mutation in the SucA enzyme, part of the 2-oxoglutarate dehydrogenase complex, increased antibiotic resistance by preventing the antibiotic-mediated induction of tricarboxylic acid (TCA) cycle activity, avoiding metabolic toxicity, and minimizing lethality [6]. These metabolic mutations were identified in clinical E. coli pathogens at levels comparable to known resistance mutations, indicating their clinical relevance [6]. It should be noted that TCA cycle activity is known to influence bacterial susceptibility to antibiotics, and its modulation has been linked to both antibiotic resistance and tolerance. The inactivation of the TCA cycle resulted in enhancing persister cell formation in S. aureus [7], and down-regulation of the TCA cycle was related to levofloxacin resistance in Vibrio alginolyticus [8]. In addition, increasing the TCA cycle flux can promote aminoglycoside uptake, thereby eliminating the drug-resistant Gram-negative bacteria [9][10].
2. Identified Key Genes Conferring Cross-Resistance and Collateral Sensitivity in E. coli
The comprehensive high-throughput laboratory evolution of E. coli systematically investigated the underlying mechanisms of cross-resistance and collateral sensitivity [3]. The key genes associated with these phenomena in E. coli are cataloged in Table 1. Notably, the study emphasized the pivotal role of mutations in genes governing transporters and porins in mediating antibiotic resistance in E. coli [3]. Perturbations in uptake and efflux activities emerged as principal mechanisms governing cross-genetic resistance and heteroresistance [3]. Specifically, the study illuminated the significance of the overexpression of efflux pumps AcrAB/TolC and EmrAB/TolC, coupled with the inactivation of their repressors, in conferring resistance to a spectrum of antibiotics [3]. Furthermore, the investigation pinpointed the involvement of YcbZ, a putative protease implicated in translation and ribosome biogenesis, in mediating cross-resistance against multiple antibiotics [3]. Mutations in PrlF, associated with the PrlF-YhaV toxin–antitoxin system, were found to be linked to resistance against specific antibiotics, such as aztreonam and carbenicillin [3]. The observed cross-resistance was partly ascribed to the diminished expression of OmpF, underscoring the intricate interplay of genes in the stress response [3]. Additionally, the study uncovered collateral sensitivities tied to prlF-mediated resistance, revealing trade-offs between acquired resistances and susceptibility to specific drugs, including rifampicin [3]. It was postulated that these collateral sensitivities might be linked to the global mRNA destabilization effect induced by increased RNase activity of PrlF [3]. Moreover, the derepression of an alternative sigma factor, RpoS, resulting from a mutation in the regular RssB, conferred both cross-resistance and collateral sensitivity to various drugs [3]. The proposed mechanism for such collateral sensitivity involved the competition between RpoS and the housekeeping sigma factor RpoD, leading to decreased carbon source availabilities and diminished competitiveness for low concentrations of nutrients [3].
Table 1. Catalog of key genes conferring cross-resistance and collateral sensitivity in E. coli.
| Mutation | Cross-Resistance | Collateral Sensitivity |
|---|---|---|
| ompF | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
| rssB | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, Tetracycline, Erythromycin, Puromycin | Protamine Sulfate, D-Cycloserine |
| glpT | Carbenicillin, Fosfomycin, Mitomycin C, Phleomycin, Puromycin | Protamine Sulfate, D-Cycloserine, Erythromycin |
| cyaA | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Fosfomycin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Erythromycin, Puromycin | Vancomycin |
| ycbZ | Chloramphenicol, Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | D-Cycloserine |
| cyoE | Kanamycin, D-Cycloserine, Phleomycin, DL-3-hydroxynorvaline, Puromycin | Rifampicin, Erythromycin |
| cyoA | Kanamycin, Phleomycin | Vancomycin |
| cyoB | Kanamycin, D-Cycloserine, Phleomycin | Sulfisoxazole, Vancomycin |
| nuoG | Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Chloramphenicol |
| mipA | Acriflavine, Mitomycin C, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Vancomycin |
| ptsP | Aztreonam, Kanamycin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | D-Cycloserine |
| rfe | Rifampicin, Carbenicillin, Mitomycin C, Phleomycin, Mecillinam | D-Cycloserine |
| purR | Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | Sulfisoxazole, D-Cycloserine |
| corA | Chloramphenicol, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Sulfisoxazole |
| oxyR | DL-3-hydroxynorvaline | Norfloxacin |
| apt | Carbenicillin, Puromycin | D-Cycloserine |
| sdaA | Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Puromycin | D-Cycloserine |
| nfsA | Chloramphenicol, Aztreonam, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Nitrofurantoin, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
| ilvL | Chloramphenicol, Acriflavine, Carbenicillin, Puromycin | Sulfisoxazole |
| gshA | Chloramphenicol, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | Sulfisoxazole |
| dacA | Chloramphenicol, Acriflavine, Mitomycin C, Phleomycin | Cefmetazole, Erythromycin |
| frlA | Chloramphenicol, Acriflavine | Vancomycin |
| fusA | Chloramphenicol, Rifampicin, Kanamycin, Acriflavine, Carbenicillin, Sulfisoxazole | Protamine Sulfate, D-Cycloserine, Vancomycin |
| glyT | Chloramphenicol, Aztreonam, Acriflavine, Carbenicillin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Erythromycin, Puromycin | Sulfisoxazole |
| gyrA | Chloramphenicol, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Tetracycline, Erythromycin, Puromycin | Acriflavine, Fosfomycin, D-Cycloserine |
| hisS | Chloramphenicol, Tetracycline | D-Cycloserine |
| iscR | Chloramphenicol, Carbenicillin, Mitomycin C, Puromycin | D-Cycloserin |
| livM | Chloramphenicol, Carbenicillin, Tetracycline\ | Sulfisoxazole |
| lon | Chloramphenicol, Cefmetazole, Acriflavine, Carbenicillin, D-Cycloserine, Phleomycin, DL-3-hydroxynorvaline, Mecillinam, Tetracycline, Erythromycin, Puromycin | Mitomycin C |
| rne | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Mitomycin C, Erythromycin | Sulfisoxazole, Vancomycin |
| rob | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Carbenicillin, Norfloxacin, Mitomycin C, Tetracycline, Erythromycin, Puromycin | D-Cycloserine |
| rpoB | Chloramphenicol, Carbenicillin, Mitomycin C, Amitriptyline, Tetracycline, Puromycin | D-Cycloserine |
| serA | Chloramphenicol, Aztreonam, Carbenicillin, Norfloxacin, Phleomycin, DL-3-hydroxynorvaline, Tetracycline, Puromycin | Fosfomycin, D-Cycloserine |
| prlF | Chloramphenicol, Aztreonam, Kanamycin, Carbenicillin, D-Cycloserine, Phleomycin, Mecillinam, Puromycin | Rifampicin |
| yjcO | Carbenicillin, Phleomycin, Tetracycline | Sulfisoxazole, D-Cycloserine |
| acrR | Chloramphenicol, Rifampicin, Cefmetazole, Aztreonam, Acriflavine, Carbenicillin, Mitomycin C, Tetracycline, Promethazine, Nitrofurantoin, Furaltadone, Erythromycin, Puromycin | D-Cycloserine |
3. Costs of AMR Evolution
The impact of evolution in the absence of antibiotics on the fitness effects of resistance mutations in E. coli was analyzed [11]. Rifampicin-resistant and -sensitive E. coli were subjected to experimental evolution in a drug-free environment [11]. The fitness effects of newly acquired resistance elements were quantified under antibiotic-free conditions [11]. The results revealed that streptomycin-resistance mutations exhibited smaller fitness effects in rifampicin-resistant genotypes adapted to antibiotic-free growth medium compared to non-adapted genotypes [11]. This epistatic variation in the costs of resistance was observed not only between different resistance mutations, but also between resistance elements and beneficial mutations acquired during adaptation to drug-free conditions [11].
The rates of resistance evolution in bacteria exposed to different antibiotics were analyzed by quantifying the distribution of fitness effects (DFE) of mutations [12]. The DFE of mutations refers to the range and characteristics of fitness changes that result from genetic mutations within a population. The DFE is thought to be a crucial factor influencing the evolutionary dynamics of drug resistance in bacterial populations [12]. The DFE in the presence of eight antibiotics, representing various modes of action, was comprehensively measured [12]. Surprisingly, the width of the DFE varied dramatically between antibiotics, with some drugs exhibiting a lower DFE width than that in the absence of stress [12]. This divergence in DFE width was attributed to distinct, drug-specific dose–response characteristics [12]. This research identified resistance variability and dose sensitivity as key drug-specific properties shaping the DFE [12]. Nitrofurantoin, in particular, exhibited exceptionally low resistance variability, limiting the evolution of resistance. Laboratory evolution experiments using nitrofurantoin confirmed slow resistance evolution via reproducible mutations [12].
Metabolic constraints on AMR evolution were investigated through laboratory evolution of E. coli under three different antibiotics (ampicillin, chloramphenicol, and norfloxacin) while growing on glucose or acetate as the sole carbon source [13]. Profiling more than 500 intracellular and extracellular metabolites in 190 evolved populations revealed that carbon and energy metabolism strongly constrain the evolutionary trajectories of antibiotic resistance [13]. Both the speed and mode of resistance acquisition were influenced by metabolic adaptations [13]. This study demonstrated that resistance evolves more rapidly on glucose compared to acetate, indicating greater metabolic plasticity during respiro-fermentative metabolism [13]. Environmental conditions played a crucial role in determining the trade-off between the costs and benefits of resistance mutations, influencing how rapidly a resistant mutant establishes itself within the population. This study identified condition-dependent compensatory mechanisms in antibiotic-resistant populations, including shifts in metabolic pathways [13]. For example, there was a shift from respiratory to fermentative metabolism of glucose upon overexpression of efflux pumps, highlighting the role of metabolic adaptations in response to antibiotic selection pressure [13]. Metabolome-based predictions revealed emerging weaknesses in antibiotic-resistant strains, such as the hypersensitivity to fosfomycin in ampicillin-resistant strains [13]. Additionally, monitoring ~750 intracellular metabolites in E. coli immediately after antibiotic exposure revealed the early bacterial response to antimicrobial compounds and potential weaknesses in terms of tolerating antibiotic therapies [14]. For example, unbalanced ammonia metabolism contributed to increased chloramphenicol toxicity, linking it to the generation of reactive oxygen species [14].
The transferability of ten resistance-conferring mutations and resistance genes (acrR, envZ, fis, gyrA, mprA, ompC, phoQ, soxR, trkH, and ycbZ) between E. coli and the closely related S. enterica subsp. serovar Typhimurium strain was investigated [15]. This study revealed that the resistance mutations in E. coli often did not confer resistance in S. enterica [15]. Surprisingly, in some cases, these mutations led to drug hypersensitivity in S. enterica [15]. In-depth analysis of a key gene involved in aminoglycoside resistance (trkH) indicated that extreme differences in mutational effects between the two species were attributed to preexisting mutations in other genes [15]. This study emphasized the limited conservation of mutational effects driving collateral sensitivity and cross-resistance phenotypes among antibiotic-resistant bacteria. The effects strongly depended on the genetic background, and even a single resistance mutation could alter collateral responses to other antibiotics.
4. Impact of Multidrug Combinations on AMR Evolution
The impact of multidrug combinations on the competitive selection between sensitive and resistant bacterial populations was analyzed using doxycycline-resistant and doxycycline-sensitive E. coli [16]. In a hyper-antagonistic class of drug combinations, a drug can render the combined treatment selective against its resistance allele. This inverted selection is insensitive to the underlying resistance mechanism and occurs at sublethal concentrations while maintaining inhibition of the wild type. The findings suggest a trade-off between the effect of drug interactions on absolute potency and the relative competitive selection imposed on emerging resistant populations. The study emphasizes a previously unappreciated feature of the fitness landscape for the evolution of resistance and points to potential avenues for designing antimicrobial combinations with improved selection against resistance [16].
The dynamics of resistance development in E. coli when exposed to five different single antibiotics (ciprofloxacin, amikacin, tetracycline, chloramphenicol, and piperacillin) and all possible antibiotic drug pairs were investigated [17]. The degree of resistance development against drug combinations was found to be linked to collateral sensitivity and the resistance that occurred during adaptation to the component drugs [17]. It was confirmed that drug resistance causing collateral sensitivity was suppressed in the growth environment under the treatment of amikacin and the other drug pairs [17]. This implies that drug combinations can limit the evolution of resistance if resistance to one drug in the combination results in collateral sensitivity to another drug.
The evolutionary conservation of collateral responses to five clinically relevant antibiotics (cefepime, ciprofloxacin, gentamicin, meropenem, and tetracycline) across species, particularly within the ESKAPE pathogens, was investigated [18]. This study identified 14 instances of universal cross-resistance that accounted for 80% of all the collateral responses and two global collateral sensitivity relationships (cefepime and gentamicin pair and ciprofloxacin and gentamicin pair) among the evolved lineages [18]. Although the genetic basis for the collateral sensitivity was unclear, genomic analyses revealed divergent and conserved evolutionary trajectories, suggesting that collateral responses may be preserved across species for the selected drugs [18].
5. Mechanism of the Trade-Off between AMR Evolution
The trade-off relationships between aminoglycoside resistance and other drugs were demonstrated by several studies (Figure 1) [2][19]. Mutations in genes whose products are involved in respiration, such as cytochrome bo3 oxidase and NDH-I, were commonly identified in aminoglycoside-evolved strains [2][19]. Since aminoglycoside uptake is known to require proton motive force (PMF) [20][21], decreased respiration results in a reduced PMF and inhibition of aminoglycoside uptake. On the other hand, activation of the multidrug efflux pump AcrAB/TolC through the inactivation of its repressor AcrR is a common multidrug resistance mechanism against various antibiotics, except for aminoglycosides [1][2][3]. Since AcrAB/TolC is a PMF-dependent proton antiporter [22][23], a decrease in PMF leads to hyper-susceptibility to other drugs that are discharged by AcrAB/TolC. Therefore, the acquisition of simultaneous resistances against both aminoglycoside and other drugs that are discharged by AcrAB/TolC poses a significant challenge. This difficulty was experimentally demonstrated by the laboratory evolution of E. coli under the simultaneous addition of two-drug combinations [17][24].
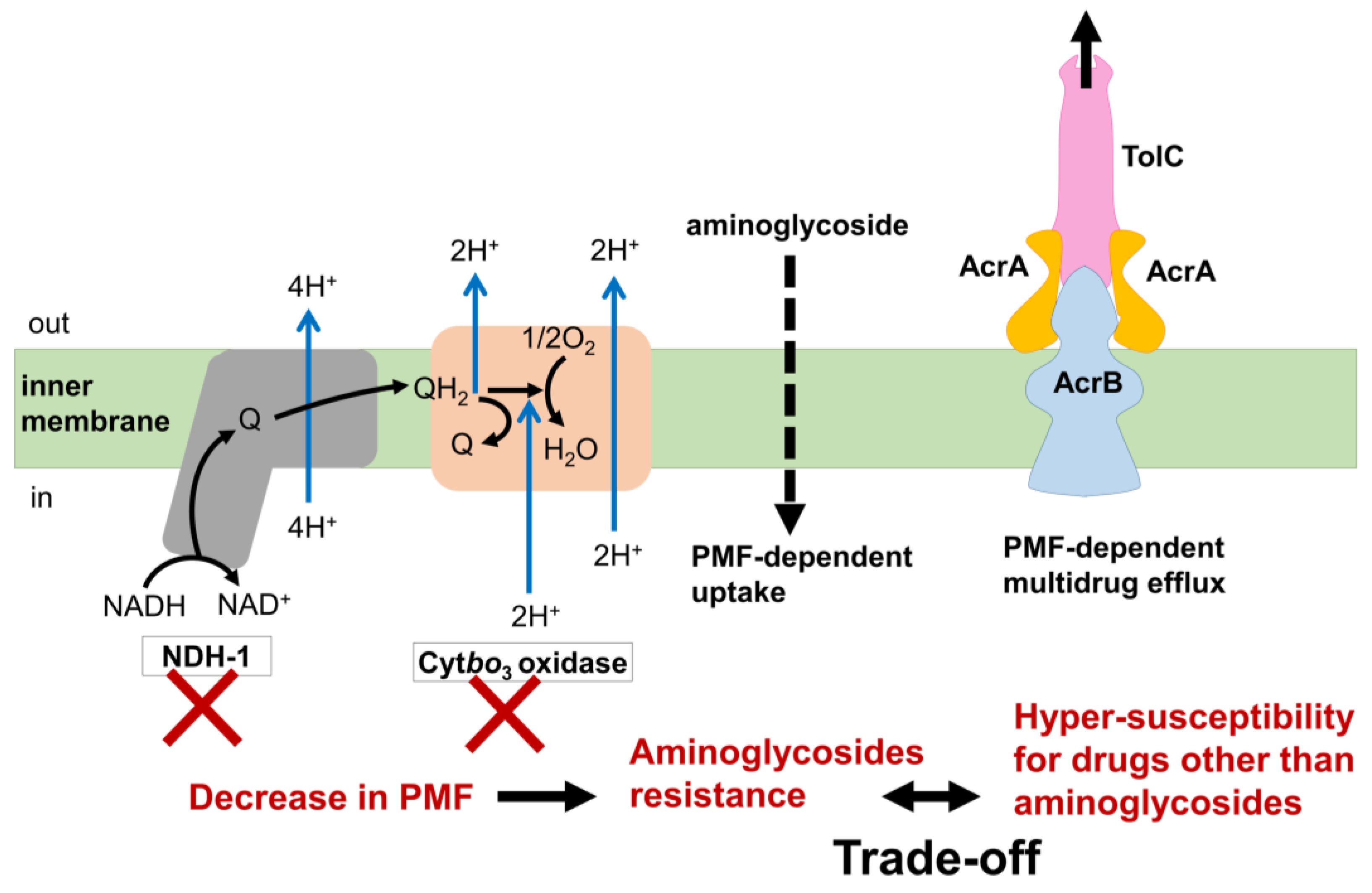
Figure 1. The trade-off mechanism of aminoglycoside resistance and drugs other than aminoglycosides. Q, quinone; QH2, quinol. E. coli possesses two isozymes with different abilities to generate PMF for NADH dehydrogenase and cytochrome oxidase in the respiratory chain. Aminoglycosides are known to be taken up into cells in a PMF-dependent manner. E. coli develops resistance to aminoglycosides through mutations that decrease PMF generation in either NDH-I or Cytbo3 oxidase, both of which exhibit a high ability to generate PMF. In contrast, many antibiotics other than aminoglycosides develop resistance by enhancing the activity of PMF-dependent multidrug efflux systems like AcrAB/TolC. However, this resistance mechanism requires a high PMF. Therefore, a trade-off occurs between resistance to aminoglycosides and resistance to other drugs due to the contrasting PMF requirements in these two resistance pathways.
6. Population Dynamics of AMR Evolution
The population dynamics of norfloxacin resistance were analyzed in a bioreactor [25]. The majority of individual evolved isolates were less resistant, with lower MICs than the group MIC. Although highly resistant isolates with higher MICs than the bioreactor concentration were rare, their presence was notable [25]. The highly resistant mutants were often present in low abundance in the population, and their emergence preceded increases in the group MIC [25]. The beneficial effect of the highly resistant mutants on the major lower resistant mutants resulted from indole production [25]. The highly resistant mutants endured a fitness cost to produce and share indole, acting as a form of altruism [25]. The production of indole by highly resistant mutants served to turn on drug efflux pumps and oxidative-stress-protective mechanisms, enhancing the survival of less resistant isolates [25]. This altruistic behavior, despite imposing a fitness cost on the highly resistant mutants, allowed weaker constituents to survive antibiotic stress [25].
7. AMR Evolution in Spatially Structured Environments
The spatial dynamics of bacterial evolution, emphasizing the interplay between mutational diversity, spatial constraints, and the ultimate fitness of the evolving population, were investigated using a growth arena (MEGA)-plate, a large experimental device allowing for the study of evolution in a large, spatially structured environment [26]. Mutants with high resistance to trimethoprim or ciprofloxacin did not always lead the evolutionary front. Highly resistant mutants could be trapped behind more sensitive lineages [26]. The physical blocking of mutants by each other was observed, reminiscent of phenomena observed in biofilm formation [26]. Mutations that increased resistance often came at the cost of reduced growth, which was restored by compensatory mutations [26]. Compensatory mutants were spatially restricted, appearing in localized spots behind the evolving front [26]. These mutants, though able to outcompete their parent in certain conditions, were constrained from contributing to the ultimate evolutionary course of the population when spatially restricted [26]. The fitness of the bacterial population was found to be determined not only by the fittest mutants, but also by those that were sufficiently fit and arose sufficiently close to the advancing front [26]. Mutants with enhanced resistance and compensatory mutations, when appearing at the front without being physically blocked, accelerated the adaptive process [26].
8. Fitness Landscape and AMR Evolution
The fitness landscape represents the relationship between genotype or phenotype and fitness in the context of antibiotic resistance evolution [27]. Since the fitness landscape is crucial for explaining and predicting evolutionary trajectories, the phenotype-based fitness landscape for AMR evolution in E. coli was investigated by quantifying the multidimensional phenotypic changes, i.e., time-series data of resistance for eight different drugs [27]. Because of the challenge regarding the high dimensionality of genotypic changes, this study quantified multidimensional phenotypic changes using time-series data on resistance for eight different drugs. This study reveals that different peaks in the phenotype-based fitness landscape correspond to different drug resistance mechanisms [27]. This finding supports the validity of the inferred landscape as a representation of the relationship between phenotype and fitness in the context of AMR [27]. A distinctive aspect of the study is the empirical approach used to infer the fitness landscape based on antibiotic resistance profiles rather than genotypes. The immense number of possible genotype changes associated with AMR makes empirical fitness landscapes based on genotypes less capable of predicting and controlling evolution. This study demonstrates that the directions of evolution predicted by the inferred phenotype-based fitness landscape align with the observed experimental trajectories, at least for evolution under certain antibiotics (tetracycline, kanamycin, and norfloxacin) [27]. This consistency suggests that resistance profiles capture the internal degrees of freedom of E. coli for predicting evolution.
9. Antibiotic Tolerance and Persistence Development
It has been demonstrated that conditions inducing bacterial tolerance can operate in various non-specific ways, both in vitro and during infection [28][29][30][31][32]. While tolerant populations exhibit high survival rates under antibiotic treatment, they suffer from impaired proliferation during infection. In contrast, antibiotic persistence is highlighted as a risk-limiting strategy, enabling bacteria to survive antibiotic treatment without compromising their ability to colonize the host [28]. The findings emphasize the importance of understanding the dynamics of these recalcitrance mechanisms to improve the design of more effective antibiotic therapies, considering the different fitness trade-offs associated with tolerance and persistence.
Metabolic homeostasis, the balance of metabolic processes in the cell, can be perturbed to promote antibiotic persistence [33][34][35][36][37][38][39][40][41]. A laboratory evolution and population-wide sequencing identified mutations in respiratory complex I (type I NADH dehydrogenase; NDH-I) as key contributors to increased persister formation in E. coli [42]. This finding was consistent across both model and pathogenic E. coli strains [42]. Mutations in NDH-I that compromise proton pumping led to significant cytoplasmic acidification upon metabolic perturbations [42]. The proposed mechanistic model suggests that strong metabolic perturbations, such as entering the stationary phase or abrupt nutrient shifts, result in cytoplasmic acidification [42]. Mutations in NDH-I exacerbate this acidification, acting as central signaling hubs connecting perturbed metabolic homeostasis with persister cell formation [42].
References
- Lázár, V.; Nagy, I.; Spohn, R.; Csörgő, B.; Györkei, Á.; Nyerges, Á.; Horváth, B.; Vörös, A.; Busa-Fekete, R.; Hrtyan, M.; et al. Genome-Wide Analysis Captures the Determinants of the Antibiotic Cross-Resistance Interaction Network. Nat. Commun. 2014, 5, 4352.
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of Antibiotic Resistance by Gene Expression Profiles. Nat. Commun. 2014, 5, 5792.
- Maeda, T.; Iwasawa, J.; Kotani, H.; Sakata, N.; Kawada, M.; Horinouchi, T.; Sakai, A.; Tanabe, K.; Furusawa, C. High-Throughput Laboratory Evolution Reveals Evolutionary Constraints in Escherichia coli. Nat. Commun. 2020, 11, 5970.
- Band, V.I.; Hufnagel, D.A.; Jaggavarapu, S.; Sherman, E.X.; Wozniak, J.E.; Satola, S.W.; Farley, M.M.; Jacob, J.T.; Burd, E.M.; Weiss, D.S. Antibiotic Combinations That Exploit Heteroresistance to Multiple Drugs Effectively Control Infection. Nat. Microbiol. 2019, 4, 1627–1635.
- Nicoloff, H.; Hjort, K.; Levin, B.R.; Andersson, D.I. The High Prevalence of Antibiotic Heteroresistance in Pathogenic Bacteria Is Mainly Caused by Gene Amplification. Nat. Microbiol. 2019, 4, 504–514.
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically Relevant Mutations in Core Metabolic Genes Confer Antibiotic Resistance. Science 2021, 371, eaba0862.
- Wang, Y.; Bojer, M.S.; George, S.E.; Wang, Z.; Jensen, P.R.; Wolz, C.; Ingmer, H. Inactivation of TCA Cycle Enhances Staphylococcus Aureus Persister Cell Formation in Stationary Phase. Sci. Rep. 2018, 8, 10849.
- Cheng, Z.-X.; Yang, M.J.; Peng, B.; Peng, X.; Lin, X.; Li, H. The Depressed Central Carbon and Energy Metabolisms Is Associated to the Acquisition of Levofloxacin Resistance in Vibrio Alginolyticus. J. Proteomics 2018, 181, 83–91.
- Su, Y.-B.; Peng, B.; Li, H.; Cheng, Z.; Zhang, T.; Zhu, J.; Li, D.; Li, M.; Ye, J.; Du, C.; et al. Pyruvate Cycle Increases Aminoglycoside Efficacy and Provides Respiratory Energy in Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E1578–E1587.
- Peng, B.; Su, Y.-B.; Li, H.; Han, Y.; Guo, C.; Tian, Y.M.; Peng, X.X. Exogenous Alanine and/or Glucose plus Kanamycin Kills Antibiotic-Resistant Bacteria. Cell Metab. 2015, 21, 249–262.
- Angst, D.C.; Hall, A.R. The Cost of Antibiotic Resistance Depends on Evolutionary History in Escherichia coli. BMC Evol. Biol. 2013, 13, 163.
- Chevereau, G.; Dravecká, M.; Batur, T.; Guvenek, A.; Ayhan, D.H.; Toprak, E.; Bollenbach, T. Quantifying the Determinants of Evolutionary Dynamics Leading to Drug Resistance. PLoS Biol. 2015, 13, e1002299.
- Zampieri, M.; Enke, T.; Chubukov, V.; Ricci, V.; Piddock, L.; Sauer, U. Metabolic Constraints on the Evolution of Antibiotic Resistance. Mol. Syst. Biol. 2017, 13, 917.
- Zampieri, M.; Zimmermann, M.; Claassen, M.; Sauer, U. Nontargeted Metabolomics Reveals the Multilevel Response to Antibiotic Perturbations. Cell Rep. 2017, 19, 1214–1228.
- Apjok, G.; Boross, G.; Nyerges, Á.; Fekete, G.; Lázár, V.; Papp, B.; Pál, C.; Csörgo, B.; Barlow, M. Limited Evolutionary Conservation of the Phenotypic Effects of Antibiotic Resistance Mutations. Mol. Biol. Evol. 2019, 36, 1601–1611.
- Chait, R.; Craney, A.; Kishony, R. Antibiotic Interactions That Select against Resistance. Nature 2007, 446, 668–671.
- Munck, C.; Gumpert, H.K.; Wallin, A.I.N.; Wang, H.H.; Sommer, M.O.A. Prediction of Resistance Development against Drug Combinations by Collateral Responses to Component Drugs. Sci. Transl. Med. 2014, 6, 262ra156.
- Rodriguez de Evgrafov, M.C.; Faza, M.; Asimakopoulos, K.; Sommer, M.O.A. Systematic Investigation of Resistance Evolution to Common Antibiotics Reveals Conserved Collateral Responses across Common Human Pathogens. Antimicrob. Agents Chemother. 2021, 16, e01273-20.
- Lázár, V.; Pal Singh, G.; Spohn, R.; Nagy, I.; Horváth, B.; Hrtyan, M.; Busa-Fekete, R.; Bogos, B.; Méhi, O.; Csörgo, B.; et al. Bacterial Evolution of Antibiotic Hypersensitivity. Mol. Syst. Biol. 2013, 9, 700.
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.S. Bacterial Uptake of Aminoglycoside Antibiotics. Microbiol. Rev. 1987, 51, 439–457.
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-Enabled Eradication of Bacterial Persisters by Aminoglycosides. Nature 2011, 473, 216–220.
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-Dependent Multidrug Efflux Systems. Microbiol. Rev. 1996, 60, 575–608.
- Murakami, S.; Yamaguchi, A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Tanpakushitsu Kakusan Koso. 2003, 48, 26–32.
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Acceleration and Suppression of Resistance Development by Antibiotic Combinations. BMC Genom. 2017, 18, 328.
- Lee, H.H.; Molla, M.N.; Cantor, C.R.; Collins, J.J. Bacterial Charity Work Leads to Population-Wide Resistance. Nature 2010, 467, 82–85.
- Baym, M.; Lieberman, T.D.; Kelsic, E.D.; Chait, R.; Gross, R.; Yelin, I.; Kishony, R. Spatiotemporal Microbial Evolution on Antibiotic Landscapes. Science 2016, 353, 1147–1151.
- Iwasawa, J.; Maeda, T.; Shibai, A.; Kotani, H.; Kawada, M.; Furusawa, C. Analysis of the Evolution of Resistance to Multiple Antibiotics Enables Prediction of the Escherichia coli Phenotype-Based Fitness Landscape. PLoS Biol. 2022, 20, 18–20.
- Michaux, C.; Ronneau, S.; Giorgio, R.T.; Helaine, S. Antibiotic Tolerance and Persistence Have Distinct Fitness Trade-Offs. PLoS Pathog. 2022, 18, e1010963.
- Meylan, S.; Andrews, I.W.; Collins, J.J. Targeting Antibiotic Tolerance, Pathogen by Pathogen. Cell 2018, 172, 1228–1238.
- Windels, E.M.; Michiels, J.E.; van den Bergh, B.; Fauvart, M.; Michiels, J. Antibiotics: Combatting Tolerance to Stop Resistance. MBio 2019, 10, e02095-19.
- Liu, Y.; Yang, K.; Zhang, H.; Jia, Y.; Wang, Z. Combating Antibiotic Tolerance Through Activating Bacterial Metabolism. Front. Microbiol. 2020, 11, 577564.
- Boeck, L. Antibiotic Tolerance: Targeting Bacterial Survival. Curr. Opin. Microbiol. 2023, 74, 102328.
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 1622–1625.
- Spoering, A.L.; Vulić, M.; Lewis, K. GlpD and PlsB Participate in Persister Cell Formation in Escherichia coli. J. Bacteriol. 2006, 188, 5136–5144.
- Hansen, S.; Lewis, K.; Vulić, M. Role of Global Regulators and Nucleotide Metabolism in Antibiotic Tolerance in Escherichia coli. Antimicrob. Agents Chemother. 2008, 52, 2718–2726.
- Girgis, H.S.; Harris, K.; Tavazoie, S. Large Mutational Target Size for Rapid Emergence of Bacterial Persistence. Proc. Natl. Acad. Sci. USA 2012, 109, 12740–12745.
- Orman, M.A.; Brynildsen, M.P. Dormancy Is Not Necessary or Sufficient for Bacterial Persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–3239.
- Kotte, O.; Volkmer, B.; Radzikowski, J.L.; Heinemann, M. Phenotypic Bistability in Escherichia coli’s Central Carbon Metabolism. Mol. Syst. Biol. 2014, 10, 736.
- Gutierrez, A.; Jain, S.; Bhargava, P.; Hamblin, M.; Lobritz, M.A.; Collins, J.J. Understanding and Sensitizing Density-Dependent Persistence to Quinolone Antibiotics. Mol. Cell 2017, 68, 1147–1154.e3.
- Shan, Y.; Brown Gandt, A.; Rowe, S.E.; Deisinger, J.P.; Conlon, B.P.; Lewis, K.; Formation, A.P.; Shan, Y.; Gandt, A.B.; Rowe, S.E.; et al. Crossm Escherichia coli. MBio 2017, 8, 1–14.
- Zalis, E.A.; Nuxoll, A.S.; Manuse, S.; Clair, G.; Radlinski, L.C.; Conlon, B.; Adkins, J.; Lewis, K. Stochastic Variation in Expression of the Tricarboxylic Acid. Am. Soc. Microbiol. 2019, 10, 01930-19.
- Van den Bergh, B.; Schramke, H.; Michiels, J.E.; Kimkes, T.E.P.; Radzikowski, J.L.; Schimpf, J.; Vedelaar, S.R.; Burschel, S.; Dewachter, L.; Lončar, N.; et al. Mutations in Respiratory Complex I Promote Antibiotic Persistence through Alterations in Intracellular Acidity and Protein Synthesis. Nat. Commun. 2022, 13, 1–18.
More
Information
Subjects:
Evolutionary Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
775
Revisions:
3 times
(View History)
Update Date:
05 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No