Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio d'Amati | -- | 4067 | 2024-01-25 15:00:51 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Addante, F.; D’amati, A.; Santoro, A.; Angelico, G.; Inzani, F.; Arciuolo, D.; Travaglino, A.; Raffone, A.; D’alessandris, N.; Scaglione, G.; et al. Mismatch Repair Deficiency in Endometrial Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/54354 (accessed on 26 July 2026).
Addante F, D’amati A, Santoro A, Angelico G, Inzani F, Arciuolo D, et al. Mismatch Repair Deficiency in Endometrial Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/54354. Accessed July 26, 2026.
Addante, Francesca, Antonio D’amati, Angela Santoro, Giuseppe Angelico, Frediano Inzani, Damiano Arciuolo, Antonio Travaglino, Antonio Raffone, Nicoletta D’alessandris, Giulia Scaglione, et al. "Mismatch Repair Deficiency in Endometrial Cancer" Encyclopedia, https://encyclopedia.pub/entry/54354 (accessed July 26, 2026).
Addante, F., D’amati, A., Santoro, A., Angelico, G., Inzani, F., Arciuolo, D., Travaglino, A., Raffone, A., D’alessandris, N., Scaglione, G., Valente, M., Tinnirello, G., Sfregola, S., Padial Urtueta, B., Piermattei, A., Cianfrini, F., Mulè, A., Bragantini, E., & Zannoni, G.F. (2024, January 25). Mismatch Repair Deficiency in Endometrial Cancer. In Encyclopedia. https://encyclopedia.pub/entry/54354
Addante, Francesca, et al. "Mismatch Repair Deficiency in Endometrial Cancer." Encyclopedia. Web. 25 January, 2024.
Copy Citation
Among the four endometrial cancer (EC) TCGA molecular groups, the MSI/hypermutated group represents an important percentage of tumors (30%), including different histotypes, and generally confers an intermediate prognosis for affected women, also providing new immunotherapeutic strategies. Immunohistochemistry for MMR proteins (MLH1, MSH2, MSH6 and PMS2) has become the optimal diagnostic MSI surrogate worldwide.
MSI
MMR deficiency
endometrial cancer
histomolecular prognostic risk assessment
immunotherapy
1. Molecular Landscape of MSI/MMRd EC
The MSI/hypermutated group, accounting for about 30% of ECs, is characterized by MSI, mostly caused by MLH1 promoter methylation, and a high mutational rate (18 × 10−6 mutations per megabase, with a high frequency of insertions and deletions), but low copy-number variations. Thus, MSI is defined as a condition of genetic hypermutability resulting from a defective DNA mismatch repair process, and the two terms are often used interchangeably [1]. MSI occurs when, during the DNA replication or in case of iatrogenic damage, frame-shift mutations (insertions or deletions) in MMR genes involve the short repetitive DNA sequences of 1–10 nucleotides (microsatellites or short tandem repeats), distributed along the genome of both coding and non-coding regions, being particularly sensitive to DNA mismatching errors, with a subsequent increased mutational burden and MMR deficiencies. MSI can be caused by somatic or germline alterations [2][3]. Somatic alterations, accounting for 85% of cases, include biallelic epigenetic MLH1 hypermethylation (in about 77% cases of sporadic endometrial cancers); downregulation of MMR genes by microRNAs; biallelic mutations; one somatic mutation and LOH; and secondary epigenetic MSH6 silencing induced by neoadjuvant RT/CHT. Germline mutations, accounting for 5% of cases, involve MMR genes and can determine two different types of clinical syndrome:
- -
- -
-
Lynch syndrome (LS), an autosomal dominant disorder characterized by the occurrence of multiple cancers, resulting from constitutional germline mutations, affecting the DNA MMR genes MLH1, MSH2, MSH6 and PMS2; constitutional MLH1 hypermethylation; or deletion of the stop codon (3′ end truncating) of the EPCAM gene causing the epigenetic silencing of the neighboring MSH2.
Generally, the MLH1 variant is correlated with the highest risk of colorectal cancer, while the MSH2 variant is correlated with the highest risk of other cancers [6]. ECs occurring in this setting represent 3–5% of cases and, even if they may occur at any age, they often arise in young women (45–55 years). EC is the index cancer in slightly more than 50% of cases. The most common target genes that harbor MSI in endometrial cancer include TP53, FBXW7, CTNNB1, ARID1A, PIK13CA, PIK3RI, PTEN, RPL22, PTEN, KRAS, ATR, CHK1, CDC5, Caspase 5, BAX gene, and JAK1 mutations. The lifetime risk of developing EC in LS patients is up to 71%; therefore, the detection of LS in patients affected by EC is crucial for genetic counseling and for the early diagnosis of secondary malignancies. According to the 2014 Clinical Practice Statement proposed by the Society of Gynecologic Oncologists, systematic clinical screening, including personal and family history and molecular/IHC screening, should be performed in all women diagnosed with EC [7][8].
The identification of MMR pathogenic variants in germline sequencing is the gold standard for the diagnosis of LS. However, the first step for LS screening in EC is represented by immunohistochemistry. In this regard, most of the ECs with MSI/MMR deficiency shows MLH1 and PMS2 loss related to sporadic MLH1 promoter hypermethylation (met-ECs) [9][10]; the remaining MMRd EC cases may be related to LS (mut-EC9). If the loss of MLH1 is detected by immunohistochemistry, testing for the presence of MLH1 promoter hypermethylation should be performed in order to detect sporadic MLH1 loss unrelated to LS [11]. Moreover, interpretation of the loss of MLH1 expression should be performed with caution:
- -
-
Homozygous MLH1 promoter hypermethylation is predominantly associated with sporadic cases;
- -
-
Heterozygous signature of the MLH1 promoter hypermethylation, as a second-hit event results in the loss of expression of the wild-type allele in LS tumors;
- -
-
The MLH1 pathogenic variant can be associated with MLH1 promoter hypermethylation.
The presence of MLH1 promoter hypermethylation should not rule out de facto the possibility of an LS diagnosis. MLH1 promoter methylation is known to be an aging-related event, thus for early-onset cancer or in case of familial history of EC, molecular testing should be performed regardless of the MLH1 promoter hypermethylation. In cases where sporadic MLH1 hypermethylation is excluded, patients are then referred for genetic counseling and germline genetic testing to confirm the diagnosis of LS. Finally, in the absence of a germline mutation, somatic mutations have also been investigated.
Finally, in up to 59% of patients displaying MSI and/or MMRd (‘suspected LS’—sLS), germline variants affecting function or promoter hypermethylation of the MLH1 gene cannot be detected. In tumors with unexplained MMRd/MSI/MLH1-unmethylated tumors, POLE/POLD1 germline and somatic screening may serve as a marker for the sporadic origin of the disease. It is important to recognize that the presence of POLE EDM may be a novel alternative pathway of MSI in ECs, generally somatic, but it does not exclude the possibility of germline MMR mutation.
2. The Histo-Molecular Approach
2.1. MMR Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: The Relationship with Molecular and Histological Subtypes
Endometrial cancer management has greatly benefited from histopathological classification, based on the histological subtype and tumor grade of differentiation, which has allowed for prognostic stratification into discrete risk categories and guided adjuvant and surgical therapy. Low-grade (G1–G2) endometrioid endometrial carcinomas (EEC) represent the subset with the most favorable outcome. High-grade (G3) endometrioid endometrial carcinoma has demonstrated an intermediate prognosis. All the other non-endometrioid subtypes are considered as high-grade (G3) and display an aggressive behavior. This group includes many histological subtypes, some long-known, such as serous endometrial carcinomas (SEC), clear cell endometrial carcinoma (CCEC) and mixed endometrial carcinoma (MEC), but also others recently classified as undifferentiated/dedifferentiated endometrial carcinoma (UEC/DEC) and uterine carcinosarcoma (UCS). This group also includes rarer subtypes, such as neuroendocrine endometrial carcinoma (NEEC), mesonephric-like endometrial carcinoma (MLEC), and gastric/gastrointestinal-type endometrial carcinoma (GTEC) [12]. Additional significant histopathological prognostic markers have been utilized to adjust for risk, particularly in endometrial cancer (EEC), such as myometrial infiltration and lymphovascular space invasion (LVSI) [13]. Unfortunately, the pathologic evaluation alone, even though playing a fundamental role in prognostic stratification, has shown some limits, such as the imperfect reproducibility of grading determination, frequent histological overlapping between subtypes and suboptimal interobserver agreement (particularly among high-grade subtypes) [14]. The TCGA classification in four molecular groups (POLE/ultramutated; MSI/hypermutated; copy-number low/endometrioid; copy-number high/serous-like) provided novel revolutionary insights into risk stratification, discovering new predictive and prognostic biomarkers and allowing a more precise characterization of patients’ outcomes [15]. The POLE/ultramutated group is defined by somatic mutations in the exonuclease domain of DNA polymerase epsilon (POLE) and is characterized by a very high mutation rate (232 × 10−6 mutations per megabase). This group includes both low-grade and high-grade EECs, all showing an excellent prognosis and no recurrence, independent from the FIGO grade. The MSI/hypermutated group is defined by microsatellite instability and shows a high mutational rate (18 × 10−6 mutations per megabase). Similarly, this group includes both low-grade and high-grade EECs, comprehensively presenting an intermediate prognosis. The copy-number low/endometrioid group presents no specific mutations, being characterized by the absence of POLE, MMR and TP53 mutations and a low degree of somatic copy-number alterations (SCNA). This group mainly includes EECs and shows an intermediate overall prognosis. The copy-number high/serous-like group is characterized by TP53 mutations (90% of cases) and a high SCNA. This group mainly includes SECs and shows a poor overall prognosis [15]. As demonstrated by subsequent studies, the TCGA classification may be predicted using cheaper immunohistochemical surrogates of molecular prognostic and predictive markers. In fact, the immunohistochemical assessment of p53 and MMR protein expression is used as a surrogate for the identification of the copy-number-high/serous-like group and MSI/hypermutated groups, respectively. Unfortunately, a reliable surrogate of POLE sequencing has not yet been identified. However, the surrogate classification defines four groups reflecting the TCGA molecular groups: POLE-mutated (POLEmut, surrogate of POLE/hypermutated), MMR-deficient (MMRd, surrogate of MSI/hypermutated), no specific molecular profile (NSMP, surrogate of copy-number low/endometrioid) and p53-abnormal (p53abn, surrogate of copy-number high/serous-like). According to the TCGA classification, the main histological subtypes of EC are distributed as follows: low-grade EECs (6% POLEmut, 25% MMRd, 64% NSMP, 5% p53abn); high-grade EECs (12% POLEmut, 39% MMRd, 28% NSMP, 21% p53abn); SECs (100% p53abn); CCECs (4% POLEmut, 10% MMRd, 42% NSMP, 44% p53abn); UECs/DECs (12% POLEmut, 44% MMRd, 25% NSMP, 19% p53abn); and UCSs (5% POLEmut, 7% MMRd, 14% NSMP, 74% p53abn) [16][17][18][19].
MMRd/MSI EC accounts for about 25–30% of ECs [20], showing distinctive histopathological features such as (i) lower uterine segment origin; (ii) endometrioid differentiation; (iii) severe nuclear atypia with undifferentiated component; (iv) high mitotic index; (v) high tumor-infiltrating lymphocytes (TILs) and/or peri-tumoral lymphocytes: ≥40 TIL/10HPFs, with more CD8+, CD45RO+ and PD1+ T cells at the invasive tumoral margin in mut-ECs compared with met-ECs; (vi) high morphological heterogeneity; (vii) substantial lympho-vascular space invasion (LVSI); (viii) deeper myometrial invasion; and (ix) synchronous ovarian cancer (clear cell or endometrioid variants) (Table 1).
Table 1. Histopathological features frequently encountered in MMRd/MSI endometrial carcinoma.
| Histopathological Features of MMRd/MSI Endometrial Carcinoma |
|---|
| Lower uterine segment (LUS) origin |
| Endometrioid differentiation |
| Severe nuclear atypia with undifferentiated component |
| High mitotic index |
| High tumor-infiltrating lymphocytes (TILs) and/or peri-tumoral lymphocytes (≥40 TIL/10HPFs, with more CD8+, CD45RO+ and PD1+ T cells at the invasive tumoral margin) |
| High morphological heterogeneity |
| Substantial lympho-vascular space invasion (LVSI) |
| Deeper myometrial invasion |
| Synchronous ovarian cancer (clear cell or endometrioid variants) |
Regarding the prevalence of MMRd ECs across the different histotypes of EC, undifferentiated/dedifferentiated carcinoma (UEC/DEC) is the most common MMR deficient subtype (44%) followed by neuroendocrine carcinoma (42.9%), high-grade endometrioid carcinoma (39.7%), mixed forms (33.3%), low-grade endometrioid carcinoma (24.7%), clear cell carcinoma [21] (9.8%) and carcinosarcoma [22] (7.3%). Only sporadic cases of serous carcinoma and mesonephric-like carcinomas have been reported to show MMR deficiency (Table 2).
Table 2. Prevalence of different histological types of MMRd/MSI endometrial carcinoma.
| Prevalence of Different Histotypes of MMRd/MSI Endometrial Carcinoma |
|---|
| Undifferentiated/dedifferentiated carcinoma (UEC/DEC): 44% |
| Neuroendocrine carcinoma: 42.9% |
| High-grade endometrioid carcinoma: 39.7% |
| Mixed: 33.3% |
| Low-grade endometrioid carcinoma: 24.7% |
| Clear cell carcinoma: 9.8% |
| Carcinosarcoma: 7.3% |
| Serous carcinoma (sporadic) |
| Mesonephric-like carcinoma (sporadic) |
Compared to POLEmut ECs, MMRd ECs seem to be more prognostically affected by clinicopathological variables, although not as much as NSMP ECs. The ESGO-ESTRO-ESP guidelines [13] substratify MMRd ECs into different risk groups based on pathological features, such as the depth of myometrial invasion, LVSI and histotype. On the other hand, in the MMRd molecular group, grading does not matter. The overall prognosis of MMRd ECs is intermediate across different histotypes leading to worsened outcomes (with higher risk for relapse) in early-stage, low-grade EECs, intermediate prognosis in high-grade EECs and improved outcomes in non-endometrioid carcinomas (NECs) [23].
Interestingly, a recent study focused on EECs characterized by a distinctive myometrial pattern of invasion, namely the microcystic elongated, angulated and fragmented (MELF) pattern, has shown a higher frequency of MSH2-MSH6 loss in this group (7.14% in MELF+ vs. 3.96% of MELF-), suggesting a possible different molecular signature among cases with and without the MELF pattern of invasion. Moreover, as described in this study, MMR deficiency could affect the risk of nodal metastases for tumors of the same size in the MELF- population, but not in MELF+ ECs [24].
Considering that the rare MMRd serous ECs or MMRd ECs with serous features have a prognosis comparable to MMRd EECs, a similar management seems to be necessary. Different studies [21][22] describe different percentages of MMRd CCECs, but the MMRd signature has been more frequently described in mixed EEC and CCEC [23]. Although the ESGO/ESTRO/ESP guidelines include CCEC among the non-endometrioid subtypes, the p53abn CCEC is characterized by a poor outcome, while the MMRd and NSMP outcome still needs to be more clearly defined. Regarding UEC/DECs and UCSs (in particular those ones with a UEC/DEC component), they may sometimes display a better prognosis [25]. As regards NEECs, MMRd represents the most common signature and, interestingly, the more frequent MMRd mixed EEC/NEECs seem to be prognostically similar to their EEC counterpart [26]. On the contrary, up until now, MLEC appears to not have had an MMRd signature [27]. As regards the gastro-intestinal differentiation in EECs, according to some studies they might be associated with an MMRd signature, having a poorer prognosis [28]. Instead, less is currently known as regards the prognosis and MMRd signature in the pure gastro-intestinal type of EC (GTEC).
Synthetically, the MMRd group prognosis seems to be intermediate across different histological EC subtypes. In EECs usually having a good prognosis (early-stage, low-grade), MMRd represents a risk factor for recurrence [29]. Conversely, in high-grade EECs, MMRd is associated with an intermediate prognosis [30][31]. In non-endometrioid carcinomas, which typically are considered aggressive, MMRd is a favorable prognostic factor [32][33][34][35]. The current evidence suggests that the MMRd group may be considered as an intermediate-risk group regardless of the histological subtype. An exception would be UEC/DEC, in which a loss of SWI/SNF protein expression appears to be associated with aggressive behavior even in the case of an MMRd signature [36][37].
2.2. EC Histological Subtypes and Genomic Alterations: The Relationship with Molecular Classification and MMR Deficiency
In the past decade, targeted gene and exome sequencing have also allowed researchers to uncover additional genetic alterations, specifically correlated with each histological subtype and TCGA subgroup [38]. Overall, EECs are characterized by frequent alterations of the PI3K–PTEN–AKT–mTOR, RAS–MEK–ERK and WNT–β-catenin pathways. Moreover, the ARID1A tumor suppressor gene is also frequently dysregulated [39]. In a recent study by Da Cruz Paula et al., PTEN (86%), ARID1A (66%), PIK3CA (56%), PIK3R1 (34%) and CTNNB1 (27%) were found to be the most commonly mutated genes in the endometrioid histological subtype [40]. A step-wise increment in the frequency of specific driver mutations was observed in FIGO Grade 1, Grade 2 and Grade 3 EECs, including ARID1A (54%, 80% and 90%, respectively), KMT2D (14%, 26% and 80%, respectively) and TP53 (8%, 14% and 50%, respectively) [40]. As previously discussed, a relatively high incidence of POLE mutations and a high rate of MSI, reflecting MMR protein defects, are detectable in EECs. Mutational signature analysis in EECs revealed that 80% of POLEmut cases had a dominant signature associated with POLE, while the other 20% had dominant signatures associated with aging or MMRd. Of the MMRd EECs, 68% had a dominant signature associated with MMR deficiency, whereas the remaining 32% showed a dominant signature associated with aging [40]. In this study, several differences in mutational profiles between early- and advanced-stage EECs were identified. Early-stage EECs were more likely to harbor POLE mutations and POLE signatures, but showed a lower incidence of MMRd-related mutational signatures. Moreover, early-stage EECs had a higher frequency of PTEN mutations. Conversely, advanced-stage EECs more frequently presented JAK1, ARID1B, SOX17 and MDC1 mutations. After excluding MSI-high and POLEmut cases, an even higher incidence of SOX17 alterations has been found in advanced-stage EECs [40]. In sporadic EECs, MSI is mainly due to MLH1 gene epigenetic silencing as a consequence of promoter hypermethylation. This alteration results in MMR deficiency and the accumulation of somatic mutations throughout the genome. Some of these mutations may represent pathogenic driver events. Recent studies have described ATR, CTCF, JAK1, RNF43 and RPL22 as driver genes that are frequently mutated in MMRd EECs [41][42][43][44][45]. Mutations in the TP53 gene are the most frequent molecular aberrations in serous carcinomas, occurring in >85% of the cases and representing an early pathogenetic event in this histological subtype [46][47][48]. In addition to TP53 mutations, other somatic mutations in SECs involve the PPP2R1A, FBXW7, SPOP, CHD4 and TAF1 genes; ERBB2, MYC and CCNE1 amplifications and p16 and synuclein-γ overexpression have also been described. The druggable PI3K pathway may also be altered in SECs, more frequently because of PIK3CA mutations, less frequently due to PTEN or PIK3R1 mutations [38]. In the study by Da Cruz Paula et al., TP53 (94%), PPP2R1A (41%), PIK3CA (35%) and FBXW7 (18%) were the most frequently mutated genes in SECs. ERRB2 alterations (hotspot mutations and amplification) and CCNE1 amplification were observed in 29% and 18% of SECs, respectively. In this study, no differences in mutations and copy-number alterations have been found between early-stage and advanced-stage SECs. However, a numerically higher frequency of ERBB2 amplification were observed in advanced-stage SECs [40]. CCECs were not included in the histological subtypes analyzed by TCGA; therefore, the molecular features of this subtype remain less studied in comparison with EECs or SECs. However, in the studies currently reported in the literature, TP53 has been found mutated in 31–50% of cases. MSI and abnormal MMR protein expression have been detected in 0–19% of cases. Other described mutations regard PPP2R1A (16–32%), PIK3CA (14–37%), FBXW7 (7–27%), PTEN (0–25%), KRAS (0–13%), ARID1A (14–22%), SPOP (14–29%) and POLE (0–6%). Additionally, genomic gains have been described for CCNE1 (18%), ERBB2 (11%) and CEBP1 (11%), whereas deletions have been reported for DAXX (11%) [33][49][50][51][52][53]. As regards UCSs, TP53 represents the most commonly mutated gene (64–91%). Other frequent mutations regard FBXW7 (11–38%), PTEN (18–47%), PIK3CA (15–41%), CHD4 (16–17%), ARID1A (10–24%), KRAS (9–29%), PPP2R1A (13–27%) and FOXA2 (5–15%). Other genes that are putative drivers of uterine carcinosarcoma are RB1 (4–11%), U2AF1 (4%), ZBTB7B (11%), ARHGAP35 (11%), SPOP (7–18%), HIST1H2BJ (7%) and HIST1H2BG (7%). Interestingly, RB1, U2AF1, and ZBTB7B are considered to be driver genes in UCSs but not in SECs or EECs. Moreover, a copy-number gain on chromosome 5p, including the TERT cancer gene, is more frequently present in UCSs compared to other histological subtypes (50% versus 17%, respectively). POLE mutations have been found in only 2–4% of UCSs. MSI has been observed in a variable percentage of cases (3.5–21%) [34][35][54][55][56][57][58][59]. A recent study by Asami et al. analyzed 1029 patients with endometrial cancer, investigating different genetic alterations in the four molecular subtypes and correlating them with prognosis [60]. TP53 mutations were significantly more common in the p53abn group than in the other three groups. PTEN and ARID1A mutations were significantly less common in the p53abn group compared to the other groups. KRAS mutations were found more frequently in the NSMP group. No gene mutations were found to be more frequently associated with the MMRd group [60].
2.3. MMR Deficiency in Light of 2021 ESGO-ESTRO-ESP Guidelines and 2023 FIGO Staging System: A Combined Histo-Molecular Approach for Risk Stratification
Figure 1 shows a diagrammatic representation and an algorithmic approach of how MMRd and the other molecular groups may influence the outcome, when combined with histological subtype and clinicopathological variables, according to the ESGO-ESTRO-ESP risk groups.
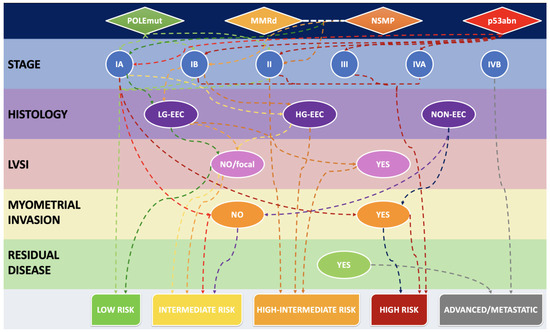
Figure 1. Algorithmic approach to stratify the risk, starting from the molecular group and combining it with staging, histological subtype and other relevant clinicopathological features. LG-EEC: low-grade endometrioid endometrial carcinoma. HG-EEC: high-grade endometrioid endometrial carcinoma. NON-EEC: non-endometrioid endometrial carcinoma.
The updated 2023 FIGO staging of EC combined molecular classification and the various histological types to better reflect the complex nature of endometrial carcinomas and their biological behavior [61]. Together with molecular classification, and perhaps even more importantly, histopathological features play the central role in the 2023 FIGO staging of EC. Histological subtype is an important prognostic factor. In this revised FIGO staging, histological subtypes are divided into non-aggressive (i.e., low-grade EECs), and aggressive histological types (i.e., high-grade EECs, SECs, CCECs, MECs, UECs/DECs, UCSs, mesonephric-like and gastrointestinal-type mucinous carcinomas). Notably, high-grade EEC is a prognostically, clinically and molecularly heterogenous category, and hence is the subtype which benefits the most from molecular profiling. Otherwise, without molecular profiling, high-grade EECs cannot be included into a specific risk group. Specifically, POLEmut high-grade EECs show an excellent prognosis, and p53abn high-grade EECs have a bad prognosis. Conversely, it has been demonstrated that, irrespective of grading, the MMRd group have an intermediate prognosis, whereas NSMP high-grade EECs, particularly if estrogen receptor-negative, always display a bad prognosis [62][63]. The new 2023 FIGO staging system for EC, based on combined molecular and histological findings, is summarized in Table 3.
Table 3. 2023 FIGO Staging System of endometrial carcinoma, including molecularly defined stages (in blue italic).
| 2023 Figo Stage | Defining Criteria |
|---|---|
| IA1 | non-aggressive histological type limited to the endometrium or an endometrial polyp |
| IA2 | non-aggressive histological type involving <50% myometrium, with no/focal LVSI |
| IA3 | low-grade EEC limited to the uterus and ovary |
| IAmPOLEmut | POLEmut EC, confined to the uterine corpus or with cervical extension, regardless of LVSI or histological type |
| IB | non-aggressive histological type involving ≥50% myometrium, and with no/focal LVSI |
| IC | aggressive histological type limited to the endometrium or an endometrial polyp |
| IIA | non-aggressive histological type with invasion of the cervical stroma |
| IIB | non-aggressive histological type with substantial LVSI |
| IIC | aggressive histological type with any myometrial infiltration |
| IICmp53abn | p53abn EC, confined to the uterine corpus with any myometrial infiltration, with or without cervical invasion, and regardless of LVSI or histological type |
| IIIA1 | spread to ovary or fallopian tube (except if it meets the Stage IA3 criteria) |
| IIIA2 | involvement of uterine subserosa/serosa |
| IIIB1 | metastasis or direct spread to the vagina and/or the parametria |
| IIIB2 | metastasis to the pelvic peritoneum |
| IIIC1 | metastasis to the pelvic lymph nodes (micrometastasis = IIIC1i/macrometastasis = IIIC1ii) |
| IIIC2 | metastasis to para-aortic lymph nodes up to the renal vessels, with or without metastasis to the pelvic lymph nodes (micrometastasis = IIIC2i/macrometastasis = IIIC2ii) |
| IVA | invasion of the bladder mucosa and/or the intestinal mucosa |
| IVB | abdominal peritoneal metastasis beyond the pelvis |
| IVC | distant metastasis, including metastasis to any extra- or intra-abdominal lymph nodes above the renal vessels, lungs, liver, brain or bone |
According to the 2023 FIGO staging of EC, MMRd, similar to NSMP status, does not modify the early FIGO stages (I and II). Instead, the presence of POLE mutations or TP53 alterations now modifies the FIGO stage. As regards Stage I and II tumors, POLEmut ECs are now classified as Stage IAmPOLEmut, independent from LVSI or histological subtype. Instead, as regards p53abn tumors with the same features, they are directly upstaged and classified as Stage IICmp53abn. In the case of multiple classifiers with POLEmut or MMRd and secondary p53 abnormality, tumors should be considered as POLEmut or MMRd, and staged accordingly. As regards advanced FIGO stages (III and IV), the staging is not altered by molecular characterization. However, Stage III and IV tumors belonging to the p53abn group should be reported as Stage IIImp53abn or Stage IVmp53abn, respectively, for data collection purposes. Additionally, the same has to be conducted for MMRd tumors, which should be recorded as Stage IIImMMRd or Stage IVmMMRd for data collection and, more importantly, in view of its predictive value for treatment with immune checkpoint inhibitors and the demonstrated progression-free and (preliminary) overall survival benefit [61].
3. Immunotherapy for MSI/dMMR Gynecological Cancers
Before the “immunotherapy era”, advanced-stage and recurrent/metastatic ECs have shown a limited response to cytoreductive surgery and systemic therapy. However, in the last few years, several studies have demonstrated that MSI/MMRd ECs are unlikely to respond to conservative hormonal treatment, show a high likelihood of LVSI justifying a sentinel or other nodal procedure and have a good response to RT (including just VBT in the absence of unfavorable risk factors). The efficacy of immune checkpoint inhibitors (ICI) especially in endometrial tumors showing high mutational burdens and immune cell infiltration (immunologically ‘hot’ tumors) has also been documented. In this regard, the MSI/hypermutated group represents the best candidate for immunotherapy.
In detail, when MMR proteins are deficient, the accumulation of uncorrected DNA mutations determines the expression of novel neoantigens and a high tumor mutational burden; these events produce an increased inflammatory response.
Based on these findings, several studies have reported the results of immune checkpoint inhibitors (anti-PD-L1 antibody) in EC with MSI. In detail, the Keynote-158 study [64] demonstrated the antitumor activity and improved survival of pembrolizumab with manageable toxicity in patients with previously treated, advanced MMRd/MSI-H ECs. Therefore, pembrolizumab received FDA approval for advanced ECs showing disease progression despite systemic therapy in any setting and which are not candidates for surgery or radiation. Following the significant clinical benefits demonstrated in the RUBY/ENGOT-en6/GOG-3031/NSGO Phase III trial in patients with advanced or recurrent ECs (72% and 36% decrease in the disease progression and death risk, in dMMR/MSI-H ECs and in the overall population, respectively), the anti-PDL-1 dostarlimab has been approved by the FDA for advanced MMRd ECs using a specific companion diagnostic assay (Ventana MMR Dx). The two randomized Phase III trials (ENGOT-en6/GOG-3031/RUBY and NRG-GY018/Keynote-868) have demonstrated a statistically significant and unprecedented PFS advantage with the addition of an immune checkpoint inhibitor (ICI) (dostarlimab or pembolizumab, respectively) to standard carboplatin/paclitaxel chemotherapy followed by ICI maintenance therapy in MMRd patients with a hazard ratio (HR) of 0.28 (95% confidence interval [CI] 0.16–0.5) and 0.30 (95% CI 0.19–0.48), respectively. Positive results have also been documented with other anti-PD-L1 therapies including nivolumab, atezolizumab, avelumab and durvalumab.
References
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The Therapeutic Significance of Mutational Signatures from DNA Repair Deficiency in Cancer. Nat. Commun. 2018, 9, 3292.
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268.
- Chadwick, R.B.; Pyatt, R.E.; Niemann, T.H.; Richards, S.K.; Johnson, C.K.; Stevens, M.W.; Meek, J.E.; Hampel, H.; Prior, T.W.; de la Chapelle, A. Hereditary and Somatic DNA Mismatch Repair Gene Mutations in Sporadic Endometrial Carcinoma. J. Med. Genet. 2001, 38, 461–466.
- Galuppini, F.; Opocher, E.; Tabori, U.; Mammi, I.; Edwards, M.; Campbell, B.; Kelly, J.; Viel, A.; Quaia, M.; Rivieri, F.; et al. Concomitant IDH Wild-Type Glioblastoma and IDH1-Mutant Anaplastic Astrocytoma in a Patient with Constitutional Mismatch Repair Deficiency Syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 233–239.
- Abedalthagafi, M. Constitutional Mismatch Repair-Deficiency: Current Problems and Emerging Therapeutic Strategies. Oncotarget 2018, 9, 35458–35469.
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310.
- Spinosa, D.; Acosta, T.; Wong, J.; Kurtovic, K.; Mewshaw, J.; Collins, S.; Kauff, N.; Havrilesky, L.J.; Strickland, K.C.; Previs, R.A. Universal Screening for Lynch Syndrome in Uterine Cancer Patients: A Quality Improvement Initiative. Gynecol. Oncol. 2021, 160, 169–174.
- Lancaster, J.M.; Powell, C.B.; Chen, L.-M.; Richardson, D.L.; SGO Clinical Practice Committee. Society of Gynecologic Oncology Statement on Risk Assessment for Inherited Gynecologic Cancer Predispositions. Gynecol. Oncol. 2015, 136, 3–7.
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 Promoter Hypermethylation Is Associated with the Microsatellite Instability Phenotype in Sporadic Endometrial Carcinomas. Oncogene 1998, 17, 2413–2417.
- Simpkins, S.B.; Bocker, T.; Swisher, E.M.; Mutch, D.G.; Gersell, D.J.; Kovatich, A.J.; Palazzo, J.P.; Fishel, R.; Goodfellow, P.J. MLH1 Promoter Methylation and Gene Silencing Is the Primary Cause of Microsatellite Instability in Sporadic Endometrial Cancers. Hum. Mol. Genet. 1999, 8, 661–666.
- Leenen, C.H.M.; van Lier, M.G.F.; van Doorn, H.C.; van Leerdam, M.E.; Kooi, S.G.; de Waard, J.; Hoedemaeker, R.F.; van den Ouweland, A.M.W.; Hulspas, S.M.; Dubbink, H.J.; et al. Prospective Evaluation of Molecular Screening for Lynch Syndrome in Patients with Endometrial Cancer ≤ 70 Years. Gynecol. Oncol. 2012, 125, 414–420.
- WHO Classification of Tumours Editorial Board. Female Genital Tumours. In WHO Classification of Tumours Series, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 4, Available online: https://tumourclassification.iarc.who.int/chaptercontent/34/536 (accessed on 15 December 2023).
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP Guidelines for the Management of Patients with Endometrial Carcinoma. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 12–39.
- Murali, R.; Delair, D.F.; Bean, S.M.; Abu-Rustum, N.R.; Soslow, R.A. Evolving Roles of Histologic Evaluation and Molecular/Genomic Profiling in the Management of Endometrial Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 201–209.
- Levine, D.A.; The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73.
- Kommoss, S.; McConechy, M.K.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Kommoss, F.; Grevenkamp, F.; Karnezis, A.; et al. Final Validation of the ProMisE Molecular Classifier for Endometrial Carcinoma in a Large Population-Based Case Series. Ann. Oncol. 2018, 29, 1180–1188.
- Stelloo, E.; Nout, R.A.; Osse, E.M.; Jürgenliemk-Schulz, I.J.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Nijman, H.W.; Putter, H.; Bosse, T.; et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-Stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4215–4224.
- Talhouk, A.; McConechy, M.K.; Leung, S.; Li-Chang, H.H.; Kwon, J.S.; Melnyk, N.; Yang, W.; Senz, J.; Boyd, N.; Karnezis, A.N.; et al. A Clinically Applicable Molecular-Based Classification for Endometrial Cancers. Br. J. Cancer 2015, 113, 299–310.
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A Simple, Genomics-Based Clinical Classifier for Endometrial Cancer. Cancer 2017, 123, 802–813.
- MacDonald, N.D.; Salvesen, H.B.; Ryan, A.; Iversen, O.E.; Akslen, L.A.; Jacobs, I.J. Frequency and Prognostic Impact of Microsatellite Instability in a Large Population-Based Study of Endometrial Carcinomas. Cancer Res. 2000, 60, 1750–1752.
- Travaglino, A.; Raffone, A.; Mascolo, M.; Guida, M.; Insabato, L.; Zannoni, G.F.; Zullo, F. Clear Cell Endometrial Carcinoma and the TCGA Classification. Histopathology 2020, 76, 336–338.
- Travaglino, A.; Raffone, A.; Gencarelli, A.; Mollo, A.; Guida, M.; Insabato, L.; Santoro, A.; Zannoni, G.F.; Zullo, F. TCGA Classification of Endometrial Cancer: The Place of Carcinosarcoma. Pathol. Oncol. Res. POR 2020, 26, 2067–2073.
- Leon-Castillo, A.; Horeweg, N.; Peters, E.E.M.; Rutten, T.; ter Haar, N.; Smit, V.T.H.B.M.; Kroon, C.D.; Boennelycke, M.; Hogdall, E.; Hogdall, C.; et al. Prognostic Relevance of the Molecular Classification in High-Grade Endometrial Cancer for Patients Staged by Lymphadenectomy and without Adjuvant Treatment. Gynecol. Oncol. 2022, 164, 577–586.
- Santoro, A.; Angelico, G.; Inzani, F.; Spadola, S.; Arciuolo, D.; Valente, M.; Musarra, T.; Capelli, G.; Fanfani, F.; Gallotta, V.; et al. Pathological Features, Immunoprofile and Mismatch Repair Protein Expression Status in Uterine Endometrioid Carcinoma: Focus on MELF Pattern of Myoinvasion. Eur. J. Surg. Oncol. 2021, 47, 338–345.
- Segura, S.E.; Pedra Nobre, S.; Hussein, Y.R.; Abu-Rustum, N.R.; Weigelt, B.; Soslow, R.A.; DeLair, D.F. DNA Mismatch Repair–Deficient Endometrial Carcinosarcomas Portend Distinct Clinical, Morphologic, and Molecular Features Compared With Traditional Carcinosarcomas. Am. J. Surg. Pathol. 2020, 44, 1573.
- Howitt, B.E.; Dong, F.; Vivero, M.; Shah, V.; Lindeman, N.; Schoolmeester, J.K.; Baltay, M.; MacConaill, L.; Sholl, L.M.; Nucci, M.R.; et al. Molecular Characterization of Neuroendocrine Carcinomas of the Endometrium: Representation in All 4 TCGA Groups. Am. J. Surg. Pathol. 2020, 44, 1541.
- Horn, L.-C.; Höhn, A.K.; Krücken, I.; Stiller, M.; Obeck, U.; Brambs, C.E. Mesonephric-like Adenocarcinomas of the Uterine Corpus: Report of a Case Series and Review of the Literature Indicating Poor Prognosis for This Subtype of Endometrial Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 971–983.
- Ardighieri, L.; Palicelli, A.; Ferrari, F.; Bugatti, M.; Drera, E.; Sartori, E.; Odicino, F. Endometrial Carcinomas with Intestinal-Type Metaplasia/Differentiation: Does Mismatch Repair System Defects Matter? Case Report and Systematic Review of the Literature. J. Clin. Med. 2020, 9, 2552.
- Moroney, M.R.; Davies, K.D.; Wilberger, A.C.; Sheeder, J.; Post, M.D.; Berning, A.A.; Fisher, C.; Lefkowits, C.; Guntupalli, S.R.; Behbakht, K.; et al. Molecular Markers in Recurrent Stage I, Grade 1 Endometrioid Endometrial Cancers. Gynecol. Oncol. 2019, 153, 517–520.
- Bosse, T.; Nout, R.A.; McAlpine, J.N.; McConechy, M.K.; Britton, H.; Hussein, Y.R.; Gonzalez, C.; Ganesan, R.; Steele, J.C.; Harrison, B.T.; et al. Molecular Classification of Grade 3 Endometrioid Endometrial Cancers Identifies Distinct Prognostic Subgroups. Am. J. Surg. Pathol. 2018, 42, 561–568.
- Joehlin-Price, A.; Van Ziffle, J.; Hills, N.K.; Ladwig, N.; Rabban, J.T.; Garg, K. Molecularly Classified Uterine FIGO Grade 3 Endometrioid Carcinomas Show Distinctive Clinical Outcomes But Overlapping Morphologic Features. Am. J. Surg. Pathol. 2021, 45, 421.
- Kim, S.R.; Cloutier, B.T.; Leung, S.; Cochrane, D.; Britton, H.; Pina, A.; Storness-Bliss, C.; Farnell, D.; Huang, L.; Shum, K.; et al. Molecular Subtypes of Clear Cell Carcinoma of the Endometrium: Opportunities for Prognostic and Predictive Stratification. Gynecol. Oncol. 2020, 158, 3–11.
- DeLair, D.F.; Burke, K.A.; Selenica, P.; Lim, R.S.; Scott, S.N.; Middha, S.; Mohanty, A.S.; Cheng, D.T.; Berger, M.F.; Soslow, R.A.; et al. The Genetic Landscape of Endometrial Clear Cell Carcinomas. J. Pathol. 2017, 243, 230–241.
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; Mackenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-Depth Molecular Profiling of the Biphasic Components of Uterine Carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185.
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 2017, 31, 411–423.
- Santoro, A.; Angelico, G.; Travaglino, A.; Raffone, A.; Arciuolo, D.; D’Alessandris, N.; Inzani, F.; Zannoni, G.F. Clinico-Pathological Significance of TCGA Classification and SWI/SNF Proteins Expression in Undifferentiated/Dedifferentiated Endometrial Carcinoma: A Possible Prognostic Risk Stratification. Gynecol. Oncol. 2021, 161, 629–635.
- Travaglino, A.; Raffone, A.; Gencarelli, A.; Saracinelli, S.; Riccardi, C.; Mollo, A.; Zullo, F.; Insabato, L. Clinico-Pathological Features Associated with Mismatch Repair Deficiency in Endometrial Undifferentiated/Dedifferentiated Carcinoma: A Systematic Review and Meta-Analysis. Gynecol. Oncol. 2021, 160, 579–585.
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 339–367.
- Wiegand, K.C.; Lee, A.F.; Al-Agha, O.M.; Chow, C.; Kalloger, S.E.; Scott, D.W.; Steidl, C.; Wiseman, S.M.; Gascoyne, R.D.; Gilks, B.; et al. Loss of BAF250a (ARID1A) Is Frequent in High-Grade Endometrial Carcinomas. J. Pathol. 2011, 224, 328–333.
- Da Cruz Paula, A.; DeLair, D.F.; Ferrando, L.; Fix, D.J.; Soslow, R.A.; Park, K.J.; Chiang, S.; Reis-Filho, J.S.; Zehir, A.; Donoghue, M.T.A.; et al. Genetic and Molecular Subtype Heterogeneity in Newly Diagnosed Early- and Advanced-Stage Endometrial Cancer. Gynecol. Oncol. 2021, 161, 535–544.
- Zighelboim, I.; Schmidt, A.P.; Gao, F.; Thaker, P.H.; Powell, M.A.; Rader, J.S.; Gibb, R.K.; Mutch, D.G.; Goodfellow, P.J. ATR Mutation in Endometrioid Endometrial Cancer Is Associated With Poor Clinical Outcomes. J. Clin. Oncol. 2009, 27, 3091–3096.
- Zighelboim, I.; Mutch, D.G.; Knapp, A.; Ding, L.; Xie, M.; Cohn, D.E.; Goodfellow, P.J. High Frequency Strand Slippage Mutations in CTCF in MSI-Positive Endometrial Cancers. Hum. Mutat. 2014, 35, 63–65.
- Novetsky, A.P.; Zighelboim, I.; Thompson, D.M.; Powell, M.A.; Mutch, D.G.; Goodfellow, P.J. Frequent Mutations in the RPL22 Gene and Its Clinical and Functional Implications. Gynecol. Oncol. 2013, 128, 470–474.
- Kim, T.-M.; Laird, P.W.; Park, P.J. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell 2013, 155, 858–868.
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 Is Frequently Mutated in Colorectal and Endometrial Cancers. Nat. Genet. 2014, 46, 1264–1266.
- Tashiro, H.; Isacson, C.; Levine, R.; Kurman, R.J.; Cho, K.R.; Hedrick, L. P53 Gene Mutations Are Common in Uterine Serous Carcinoma and Occur Early in Their Pathogenesis. Am. J. Pathol. 1997, 150, 177–185.
- Sherman, M.E.; Bur, M.E.; Kurman, R.J. P53 in Endometrial Cancer and Its Putative Precursors: Evidence for Diverse Pathways of Tumorigenesis. Hum. Pathol. 1995, 26, 1268–1274.
- Vermij, L.; Léon-Castillo, A.; Singh, N.; Powell, M.E.; Edmondson, R.J.; Genestie, C.; Khaw, P.; Pyman, J.; McLachlin, C.M.; Ghatage, P.; et al. p53 immunohistochemistry in endometrial cancer: Clinical and molecular correlates in the PORTEC-3 trial. Mod Pathol. 2022, 35, 1475–1483.
- Timmers, P.J.; Zwinderman, A.H.; Teodorovic, I.; Vergote, I.; Trimbos, J.B. Clear Cell Carcinoma Compared to Serous Carcinoma in Early Ovarian Cancer: Same Prognosis in a Large Randomized Trial. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2009, 19, 88–93.
- Le Gallo, M.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; Zhang, S.; Lozy, F.; Sgroi, D.C.; Bel, A.V.; Matias-Guiu, X.; Broaddus, R.R.; et al. Somatic Mutation Profiles of Clear Cell Endometrial Tumors Revealed by Whole Exome and Targeted Gene Sequencing. Cancer 2017, 123, 3261–3268.
- An, H.-J.; Logani, S.; Isacson, C.; Ellenson, L.H. Molecular Characterization of Uterine Clear Cell Carcinoma. Mod. Pathol. 2004, 17, 530–537.
- Hoang, L.N.; McConechy, M.K.; Meng, B.; McIntyre, J.B.; Ewanowich, C.; Gilks, C.B.; Huntsman, D.G.; Köbel, M.; Lee, C.-H. Targeted Mutation Analysis of Endometrial Clear Cell Carcinoma. Histopathology 2015, 66, 664–674.
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining Prognosis and Identifying Targetable Pathways for High-Risk Endometrial Cancer; a TransPORTEC Initiative. Mod. Pathol. 2015, 28, 836–844.
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational Landscape of Uterine and Ovarian Carcinosarcomas Implicates Histone Genes in Epithelial-Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243.
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.; Senz, J.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; et al. Use of Mutation Profiles to Refine the Classification of Endometrial Carcinomas. J. Pathol. 2012, 228, 20–30.
- Raffone, A.; Travaglino, A.; Raimondo, D.; Maletta, M.; De Vivo, V.; Visiello, U.; Casadio, P.; Seracchioli, R.; Zullo, F.; Insabato, L.; et al. Uterine Carcinosarcoma vs Endometrial Serous and Clear Cell Carcinoma: A Systematic Review and Meta-analysis of Survival. Int. J. Gynecol. Obstet. 2022, 158, 520–527.
- Jones, S.; Stransky, N.; McCord, C.L.; Cerami, E.; Lagowski, J.; Kelly, D.; Angiuoli, S.V.; Sausen, M.; Kann, L.; Shukla, M.; et al. Genomic Analyses of Gynaecologic Carcinosarcomas Reveal Frequent Mutations in Chromatin Remodelling Genes. Nat. Commun. 2014, 5, 5006.
- Le Gallo, M.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program; Merino, M.J.; Mutch, D.G.; Goodfellow, P.J.; Mullikin, J.C.; Bell, D.W. The FOXA2 Transcription Factor Is Frequently Somatically Mutated in Uterine Carcinosarcomas and Carcinomas. Cancer 2018, 124, 65–73.
- Biscuola, M.; Van de Vijver, K.; Castilla, M.Á.; Romero-Pérez, L.; López-García, M.Á.; Díaz-Martín, J.; Matias-Guiu, X.; Oliva, E.; Palacios Calvo, J. Oncogene Alterations in Endometrial Carcinosarcomas. Hum. Pathol. 2013, 44, 852–859.
- Asami, Y.; Kobayashi Kato, M.; Hiranuma, K.; Matsuda, M.; Shimada, Y.; Ishikawa, M.; Koyama, T.; Komatsu, M.; Hamamoto, R.; Nagashima, M.; et al. Utility of Molecular Subtypes and Genetic Alterations for Evaluating Clinical Outcomes in 1029 Patients with Endometrial Cancer. Br. J. Cancer 2023, 128, 1582–1591.
- Berek, J.S.; Matias-Guiu, X.; Creutzberg, C.; Fotopoulou, C.; Gaffney, D.; Kehoe, S.; Lindemann, K.; Mutch, D.; Concin, N.; Endometrial Cancer Staging Subcommittee; et al. FIGO Staging of Endometrial Cancer: 2023. Int. J. Gynecol. Obstet. 2023, 162, 383–394.
- Jamieson, A.; Huvila, J.; Chiu, D.; Thompson, E.F.; Scott, S.; Salvador, S.; Vicus, D.; Helpman, L.; Gotlieb, W.; Kean, S.; et al. Grade and Estrogen Receptor Expression Identify a Subset of No Specific Molecular Profile Endometrial Carcinomas at a Very Low Risk of Disease-Specific Death. Mod. Pathol. 2023, 36, 100085.
- Vermij, L.; Jobsen, J.J.; León-Castillo, A.; Brinkhuis, M.; Roothaan, S.; Powell, M.E.; de Boer, S.M.; Khaw, P.; Mileshkin, L.R.; Fyles, A.; et al. Prognostic Refinement of NSMP High-Risk Endometrial Cancers Using Oestrogen Receptor Immunohistochemistry. Br. J. Cancer 2023, 128, 1360–1368.
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability–High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
844
Revision:
1 time
(View History)
Update Date:
25 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No