Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lyudmila Parfenova | -- | 5736 | 2024-01-23 05:10:02 | | | |
| 2 | Sirius Huang | Meta information modification | 5736 | 2024-01-24 01:42:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Parfenova, L.V.; Bikmeeva, A.K.; Kovyazin, P.V.; Khalilov, L.M. Catalytic Synthesis of Terminal Alkene Dimers and Oligomers. Encyclopedia. Available online: https://encyclopedia.pub/entry/54223 (accessed on 24 July 2026).
Parfenova LV, Bikmeeva AK, Kovyazin PV, Khalilov LM. Catalytic Synthesis of Terminal Alkene Dimers and Oligomers. Encyclopedia. Available at: https://encyclopedia.pub/entry/54223. Accessed July 24, 2026.
Parfenova, Lyudmila V., Almira Kh. Bikmeeva, Pavel V. Kovyazin, Leonard M. Khalilov. "Catalytic Synthesis of Terminal Alkene Dimers and Oligomers" Encyclopedia, https://encyclopedia.pub/entry/54223 (accessed July 24, 2026).
Parfenova, L.V., Bikmeeva, A.K., Kovyazin, P.V., & Khalilov, L.M. (2024, January 23). Catalytic Synthesis of Terminal Alkene Dimers and Oligomers. In Encyclopedia. https://encyclopedia.pub/entry/54223
Parfenova, Lyudmila V., et al. "Catalytic Synthesis of Terminal Alkene Dimers and Oligomers." Encyclopedia. Web. 23 January, 2024.
Copy Citation
Dimers and oligomers of alkenes represent a category of compounds that are in great demand in diverse industrial sectors. Among the developing synthetic methods, the catalysis of alkene dimerization and oligomerization using transition metal salts and complexes is of undoubted interest for practical applications.
dimerization
oligomerization
transition metal catalysis
metal hydrides
reaction mechanisms
1. Introduction
The dimers and oligomers of alkenes belong to a broad class of compounds that are in high demand across various industrial sectors. Typically, they are used as comonomers in ethylene polymerization and serve as raw materials for the production of adhesives, surfactants, flavors, synthetic fuel additives, and more [1][2][3][4][5][6]. Alkene oligomers are synthesized catalytically using various methods, including heterogeneous acid catalysis with the use of phosphoric acid on silica, ion exchange resins, silica-aluminas, zeolites, etc., where the classical mechanism involving carbocations (carbenium pathway) is realized [6][7][8]. Another way to synthesize oligomers involves transition metal catalysis (Zr, Ti, Hf, Ni, Co, Fe, V, and Ta) in which the Cossee–Arlman mechanism is implemented [9]. Metal-catalyzed processes, such as the oligo- and polymerization of ethylene on chromium catalysts (Philips), the preparation of linear α-olefins via ethylene oligomerization on a nickel catalyst (SHOP = shell higher olefin process), the oligomerization of light alkenes into C4–C10 olefins using AlphaButol, AlphaHexol, Dimersol, and Difasol process technologies, and others, were developed to produce olefin oligomers [10][11][12][13][14]. The oligomerization of alkenes (1-butene and 1-hexene) synthesized from renewable plant raw materials to obtain jet and diesel fuels is also attracting the attention of the scientific community [6][15].
Among the developing methods with significant potential for advancement and practical implementation is the catalysis of alkene dimerization and oligomerization using Ti subgroup metal salts and complexes, which provide high reaction rates and effective control of their chemo-, regio-, and stereoselectivity. The classical heterogeneous Ziegler–Natta catalysis is widely used in the production of polyethylene and polypropylene [16][17][18][19][20][21]. The discovery of metallocenes [22], along with organoaluminum [23][24][25] and boron activators [22][26][27], enabled the process to be transferred from a heterogeneous medium to a homogeneous one, which provided undoubted positive effects—such as an increase in the activity and the possibility to effectively control stereoselectivity—and allowed for a detailed study to be conducted on the reaction mechanisms. Homogeneous systems effectively catalyze alkene di-, oligo-, and polymerization [1][28][29][30], as well as the hydro-, carbo-, and cyclometalation of olefins and acetylenes [31][32][33][34][35], which can not only be considered as the methods of multiple bond functionalization but also as the initial stages of chain growth in the oligo- and polymerization processes.
Information concerning the catalytic alkene oligomerization and the types of products it produces can be seen in Scheme 1. The nature of the active reaction centers determines the structural types of the resulting oligomers and the regio- and stereoselectivity of a substrate’s insertion. In the processes presented in Scheme 1, hydride, alkyl, or alkene intermediates act as active reaction centers, which, in the early stages, facilitate alkene hydro-, carbo-, or cyclometalation, respectively. Chain termination occurs through the elimination of the oligomeric product, generating metal hydrides, or through the transfer of the growing chain to an organometallic cocatalyst or alkene. Metal hydrides, therefore, can serve as dominant reaction centers in these catalytic systems.
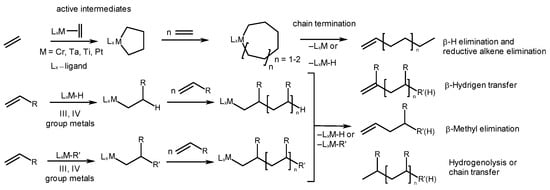
Scheme 1. Alkene oligomerization catalyzed with transition metal complexes: active centers and types of products.
Recent reviews on catalytic ethylene, propylene, and higher olefin oligomerization discussed the chemical methodologies, possible reaction mechanisms, techniques used to study the structure and physicochemical properties of oligomers, the practical implementation of these processes in the industry, and the potential applications for the resulting products [1][3][36][37][38][39][40][41][42].
2. Catalytic Synthesis of Terminal Alkene Dimers and Oligomers
In the literature, significant attention is given to Ti subgroup metal complexes, the use of which, in homogeneous catalytic systems, ensures alkene dimerization, oligomerization, and polymerization with high yields and high chemo- and stereoselectivity [1][5][40]. The selective transformation of α-olefins (propene (1a), 1-butene (1b), 1-hexene (1c), 1-octene (1d), and 3-methyl-1-butene (1e)) into vinylidene dimers (2a–e) under the action of a catalytic system consisting of Cp2ZrCl2 (3) or Cp2ZrMe2 (4) and aluminoxane, obtained in situ through the reaction of AlR3 (R = Me, Et, and Bui) with CuSO4·5H2O, was reported in one of the first works on this topic (Scheme 2) [43]. Dimeric products were obtained with a selectivity of up to 96% during the reaction performed at 40–70 °C for 0.5–2 h with the following reagent ratio: [Zr]:[Al]:[1-alkene] = 1:(8–100):(600–4670). The highest alkene conversion rate and selectivity towards the dimerization were achieved in the reaction, which was catalyzed with Cp2ZrCl2 and methylaluminoxane (MAO).
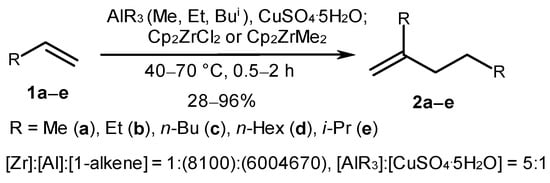
Scheme 2. Alkene dimerization under the action of a catalytic system: Cp2ZrCl2 (Cp2ZrMe2)-AlR3 (R = Me, Et, and Bui)-CuSO4·5H2O [43].
A dimeric product (2d) was obtained with a yield of 59% in the reaction of 1-octene with AlMe3 in the presence of Cp2ZrCl2 (3) in 1,2-dichloroethane at 22 °C for 12 h (Scheme 3) [44]. It was assumed that the initial stage of the reaction was the alkene carbometalation and the formation of metal alkyl 7, which hydrometalates 1-octene through state 8. As a result, 2-methyl-1-octene and the hydrometalation product, n-OctMLn, are accumulated in the mixture. The latter reacts with 1-octene via carbometalation to provide 2-(n-hexyl)-1-decyl metal that hydrometalates 1-octene to form n-OctMLn and dimer 2d (Scheme 3).
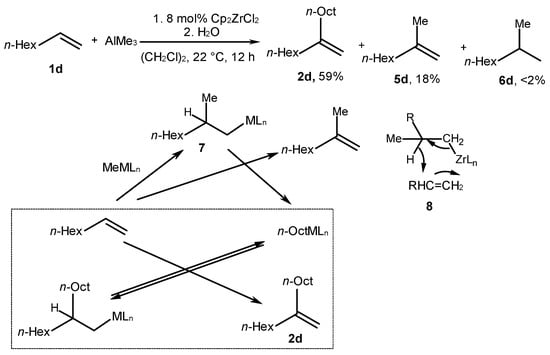
Scheme 3. Reaction of 1-octene with AlMe3 in the presence of Cp2ZrCl2 in 1,2-dichloroethane and probable mechanism [44].
Furthermore, terminal alkenes 1b, 1c, and 1e–h were dimerized in the presence of the catalytic system Cp2ZrCl2-MAO at an Al/Zr ratio of 1:1 and a room temperature of 25 °C for 24 h with the product yield of 80–90% (Scheme 4) [45][46]. 3-Methyl-1-butene (1e) provided a mixture of 2-methyl-2-butene (9e, 77%), 2-methyl-1-butene (10e, 17%), and 5-methyl-2-(methylethyl)-1-hexene (2e, 3%). The reaction of o-diallylbenzene (1i) with Cp2ZrCl2 and MAO at an Al/Zr ratio of 4:1 for 3 days provided a cyclic product, methylenecycloheptane 11i, with a 70% yield.
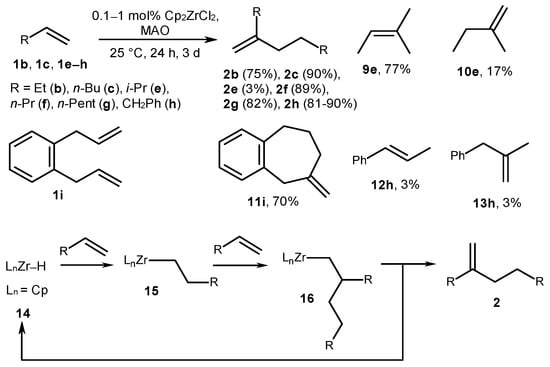
The mechanism proposed in [45][46] for the dimerization reaction implies the insertion of 1-alkene into a Zr-H bond of zirconocene hydride 14 to obtain Zr-alkyl complex 15, which then carbometalates the second alkene molecule, producing alkyl derivative 16. Subsequent β-H elimination in alkyl complex 16 provides dimer 2 and hydride complex 14 (Scheme 4). It is noted that the presence of chlorine in a catalytic system is an important factor for the dimerization reaction. In confirmation, a higher oligomer formation was seen in the presence of Cp2ZrMe2 (4) as a catalyst and MAO (without Cl atoms) was given. The chlorine atom probably ensures the fast β-H elimination, but not the growth of an alkyl chain [46].
A dimeric hydride complex, [(2,4,7-Me3-Ind)2Y(μ-H)]2 (17) (Scheme 5), appeared to be an effective catalyst for the homodimerization of various α-olefins [47]. The reaction was performed in benzene at 80–100 °C for 2–24 h, and there was a 20–50-fold molar excess of α-olefins. The head-to-tail dimerization was observed in the case of 1-hexene (1c) and 3-methyl-1-butene (1e) with a selectivity of >98%.
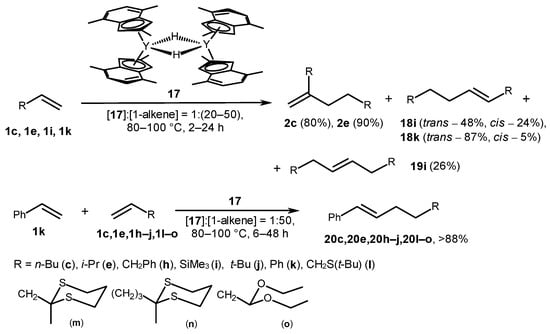
Scheme 5. α-Olefin homo- and codimerization, catalyzed with the following hydride complex: [(2,4,7-Me3-Ind)2Y(μ-H)]2 [47].
The reaction runs through sequential 1,2-insertion followed by β-H elimination (Scheme 6) [47]. The homodimerization of trimethylvinylsilane (1i) and styrene (1k) occurred, forming head-to-head products. The reactions presumably proceed through an initial 1,2-insertion into the Y-H bond, followed by a subsequent 2,1-coordination and β-H abstraction. Olefins 1l–o, containing heteroatoms, 3,3-dimethyl-1-butene (1j), and allylbenzene (1h), did not undergo homodimerization under the reaction conditions. In the reaction with 1h, C-H activation arose, resulting in the formation of a catalytically inactive allyl complex, Ind’2Y(η3-CH2CHCHPh). The reaction of 17 with 1l–o provided stable alkyl complexes that deactivated the catalyst.
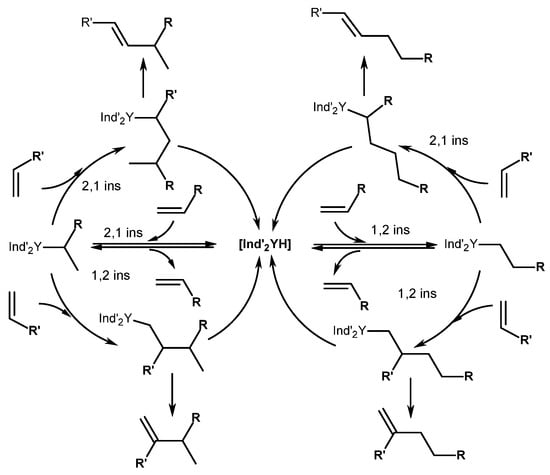
Scheme 6. Proposed mechanism of α-olefin homo- and codimerization, catalyzed with [(2,4,7-Me3-Ind)2Y(μ-H)]2 (17) [47].
Complex 17 also showed activity in the styrene codimerization with alkenes (H2C=CHR) that produced trans-1-phenyl-4-alkylbut-1-enes (20) with more than an 88% yield at 80–100 °C (Scheme 5). The reaction probably occurred through the 1,2-coordination of α-olefin into the Y-H bond, followed by a subsequent 2,1-insertion of styrene into the Y-C bond of the alkyl intermediate and β-H elimination (Scheme 6). Heteroatom-containing olefins 1l–o readily formed head-to-head codimers with styrene. However, these substrates exhibited lower reactivity, and the accompanying formation of the styrene homodimer was observed.
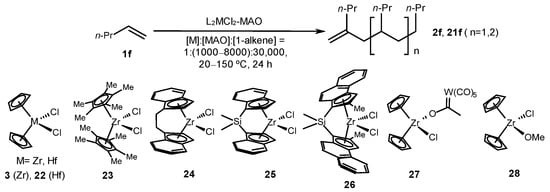
Complexes of various structures were subsequently tested in the reactions to find selective catalysts for alkene dimerization and oligomerization. For example, 1-pentene (1f) was transformed into oligomers in the presence of catalytic systems based on bis-cyclopentadienyl complexes 3, 22, and 23 and MAO in a ratio ([Zr]:[MAO]:[substrate] = 1:1000:30,000) at 60 °C for 24 h in toluene (Scheme 7) [48]. Oligomeric products with low molecular weights were obtained: dimers (25%), trimers (18%), and tetramers (14%). The use of catalysts with ansa-indenyl ligands (EBI)ZrCl2 (24) and (SBI)ZrCl2 (25) led to the formation of isotactic poly(1-pentene) (MN = 1700–4400 g mol−1, PDI = 4.75–6.41). Further studies on the catalytic properties of complexes 3 and 22–28 at a reagent ratio ([metallocene]:[MAO]:[1-alkene] = 1:(1000–8000):30,000) at 20–150 °C demonstrated the dependence of the reaction chemoselectivity upon the metallocene structure [49]. The reaction of 1-pentene, catalyzed with complexes 22 and 23 and MAO ([Zr]:[MAO] = 1:1000), resulted in an atactic polypentene, whereas ansa-indenyl complexes 24–26 provided an isotactic polymer with stereoselectivities of 0.91, 0.45, and 0.64 (mmmm), respectively. The polymer with the highest molecular weight (MW = 149,000, PDI = 1.85–2.08) was obtained by using catalyst 26. The reaction, which was catalyzed with cyclopentadienyl complexes 3, 27, and 28, under these conditions, afforded the oligomeric products with 2–4 units. In this case, the highest conversion (50%) was achieved in the presence of bimetallic complex 27, whereas the yields of dimers, trimers, and tetramers were 10, 20, and 20%, respectively. An increase in the amount of MAO to 6000 eq. in the case of complex 3 caused an increase to 80% in the alkene conversion, and the yields of dimers, trimers, and tetramers increased to 15, 30, and 35%, respectively (Scheme 7) [49].
Branched α-olefins were regioselectively dimerized at 20 °C in toluene for 3–142 h upon the action of Cp2MCl2 (M = Ti (29), Zr (3), and Hf (22)) or Me2SiCp2ZrCl2 (30) and MAO at the following ratio: [M]:[MAO] = 1:581 (Scheme 8) [50]. Complex 30 with Me2Si-bonded cyclopentadienyl ligands showed the highest activity and regioselectivity, providing dimers 2q–t with yields of up to 100%. 3-Methyl-1-butene (1e) and 3-methyl-1-pentene (1p) provided dimers with yields of 11 and 19%, as well as oligomeric products 21e and 21p, correspondingly.
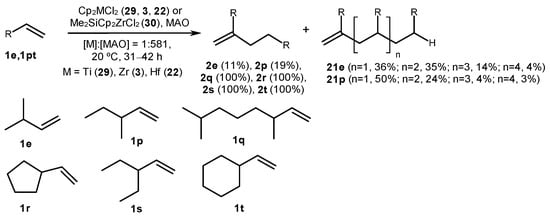
Scheme 8. Oligomerization of branched α-olefins, catalyzed with Cp2MCl2 (M = Ti (29), Zr (3), and Hf (22)) or Me2SiCp2ZrCl2 (30); yields are given for catalyst 30 [50].
As a rule, the application of other transition metal complexes changes the regioselectivity of a reaction. For example, pyridine bis(imine) iron complexes 31a,b, upon activation with MAO, MMAO, or AlR3 (R = Et, Bui)-B(C6F5)3 (Al/Fe = 70–480), demonstrate the ability to dimerize various α-olefins, such as 1b, 1c, 1u, and 1x (Scheme 9) [51]. This results in the formation of a mixture of linear olefin dimers 18b, 18c, 18u, and 18x with internal double bonds (63–80%) and monomethyl-branched dimers 35b, 35c, 35u, and 35x (18–36%). Additionally, trisubstituted vinylidene (2-alkylalkenes) or α-olefinic products were detected in trace amounts. High alkene conversion up to 76% was achieved in the presence of 31a–c and 31e at 30–50 °C. The sterically less hindered complex 31d provided monomethyl-branched dimers 35b, 35c, 35u, and 35x.
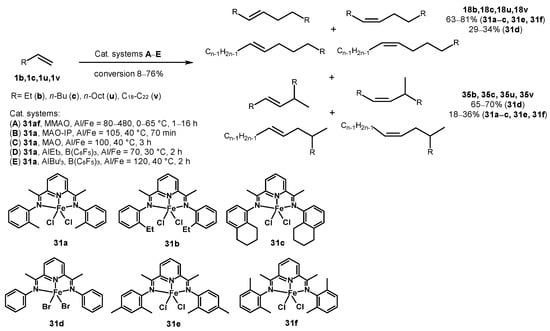
Scheme 9. Dependence of the type of alkene dimerization products on the post-metallocene structure [51].
The reaction mechanism consists of consecutive stages of the 1,2-insertion of the initial olefin into the Fe-H bond, followed by a 2,1-insertion of the second olefin (Scheme 10). Subsequent β-H elimination leads to the formation of linear dimers. Successive 2,1-insertions of alkenes and β-H eliminations produce Me-substituted dimers.
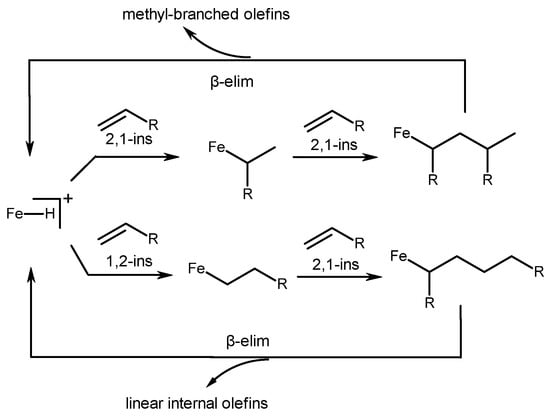
Scheme 10. Mechanism of alkene dimerization, catalyzed with iron complexes 31a–d [51].
Pyridine bis(imine) cobalt catalysts 32a–d, when activated with MMAO (Al/Co = 200–500), dimerized α-olefins with lower productivity compared to similar iron systems (TON for 1-butene: 42,000 vs. 147,000 for Co and Fe, respectively) (Scheme 11) [52]. The main products were linear dimers (>97%) and butene isomers in an 18b/iso-1b ratio of 0.47–0.7. In the dimerization of propylene, linear hexenes, nonenes, and dodecenes were obtained with turnovers exceeding 200,000 moles of propylene/mole Co (17,000 g oligomer/g Co complex). Complexes 32a and 32b, in combination with MMAO or EtAlCl2, induced the isomerization of 1-hexene.
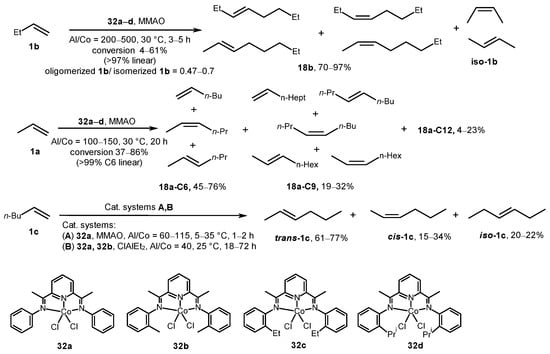
Scheme 11. Pyridine bis(imine) cobalt complexes 32a–d as catalysts of α-olefin dimerization [52].
The main stages of the stepwise reaction mechanism include a consecutive 1,2-insertion of olefin and a 2,1-insertion into Co-Alkyl, followed by chain termination to provide alkenes with internal and terminal double bonds (Scheme 12) [52].
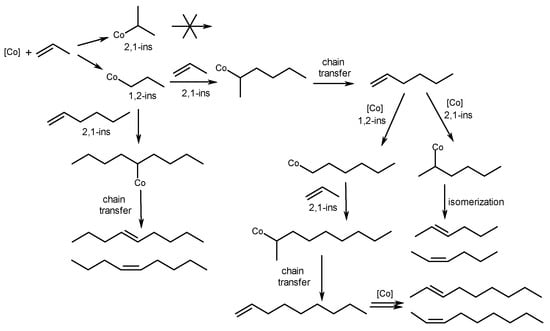
Scheme 12. Mechanism of alkene dimerization, catalyzed with Co complexes 32a–d.
Nevertheless, mixed ethylene Co complex 32e catalyzed the transformation of terminal alkenes into vinylidene dimers of a head-to-tail type with yields of 66–80% in the presence of an organoboron activator, HBArF, at a [Co]:[B]:[1-alkene] ratio of 1:0.81:670 in contrast to the post-metallocene catalysts 32a–d (Scheme 13) [53]. Moreover, linear terminal alkene 1w formed in the reaction with a yield of up to 14%, presumably due to the isomerization processes in intermediate alkyl Co complexes.
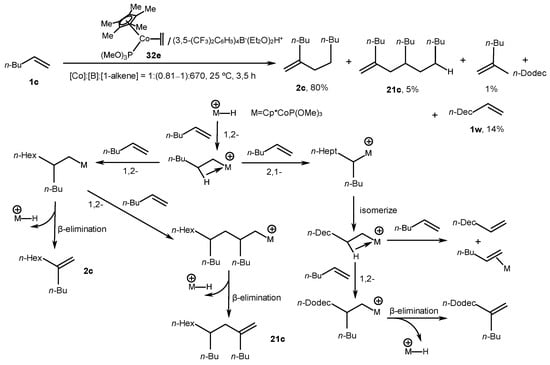
Scheme 13. Transformations of terminal alkenes into dimers, which were catalyzed with complex 32e [53].
α-Olefins 1c, 1d, 1f, 1g, and 1x undergo tail-to-tail dimerization under the action of a catalytic system, WCl6/R′NH2/R″3N/EtAlCl2, obtained in situ at a [W]:[R′NH2]:[R″3N]:[EtAlCl2]:[1-alkene] molar ratio of 1:(1–4):(0–4):12:(834–5000) to provide predominantly methyl-branched products (33c, 33d, 33f, 33g, and 33x) (Scheme 14) [54]. Alkene conversion at a level of 80% and high selectivity towards the dimerization were achieved (>99%) due to the optimal choice of a chlorine-containing organoaluminum activator (EtAlCl2) and a solvent, PhCl. This effect on the reaction initiation was attributed to the generation of bimetallic catalytically active centers with a W-Cl-Al bridge.
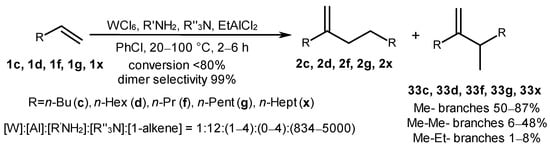
Scheme 14. Alkene dimerization under the action of a catalytic system, WCl6/R′NH2/R″3N/EtAlCl2 [54].
Upon a detailed analysis of the reaction products using the example of 1-pentene dimers, it was demonstrated that fractions of linear C10 products (constituting only 0.1% of the dimer fraction) contain trans-5-decene, cis-4-decene, dienes, and decane (Scheme 15) [54]. The authors proposed a Cossee-type mechanism [9], noting that the initial insertion of an alkene occurs equally as 1,2- and 2,1-, followed by a subsequent regioselective alkene 1,2-coordination. Therefore, the dominant structures appear to be B and C, which provide the main reaction products.
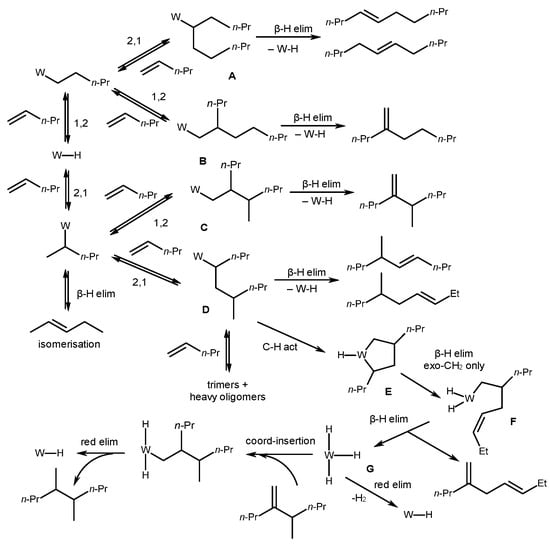
Scheme 15. Mechanism of alkene dimerization under the action of a catalytic system, WCl6/R′NH2/R″3N/Et2AlCl [54].
Low-molecular-weight oligomeric products, including 1-hexene dimers, were synthesized with high yields (73–97%) and selectivity (≥98%) in the presence of Zr and Hf post-metallocene complexes with amino-bis(phenolate) [ONNO] ligands and a neutral activator, B(C6F5)3, at 65–85 °C for 4 h and the following reagent ratio: [metallocene]:[B]:[1-hexene] = 1:1.1:100 (Scheme 16) [55]. The highest activity in the oligomerization was achieved in the presence of Zr catalysts 34a–c; in this case, the molecular weights of the products corresponded to a typical Flory–Schulz distribution [56]. Hafnium catalysts 34d–f showed lower activities in contrast to the zirconium analogs; however, they showed greater selectivity in dimerization. In addition, the molecular weight distribution of the products obtained in the presence of hafnium catalysts did not follow the Schultz–Flory distribution. The high selectivity in the formation of vinylidene dimers was explained by the prevalence of 1,2-alkene insertions into catalytically active centers, both in primary M-H and secondary M-Alkyl species. It was also noted that the chain termination rate for these systems exceeds the rate of chain propagation. In the case of regioerror, i.e., alkene 2,1-insertions, conversely, the chain propagation prevails because the elimination is practically impossible; therefore, chain termination via β-H elimination will occur when the 1,2-incorporation of an alkene takes place. The authors explain that deviations from the Schultz–Flory distribution are caused by the presence of two or more conformations of hafnium active centers, which have different activities towards alkene (the assumption was made from the 1H NMR spectra of the initial complexes depending on temperature). For zirconium analogs, it seems that either one isomer is characteristic, or the exchange between conformations is very fast (the energy barrier is small), so it does not significantly affect the distribution of oligomers.
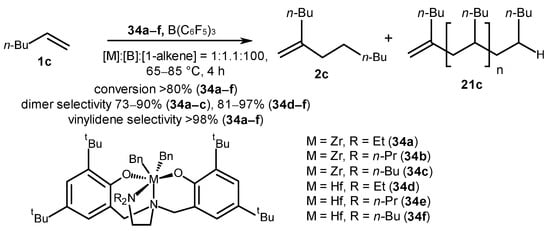
Scheme 16. Post-metallocene Zr and Hf amino-bis(phenolate) complexes of [ONNO]-type as catalysts of 1-hexene oligomerization [55].
A highly regioselective method for the oligomerization of 1-hexene and 1-octene was developed at relatively low catalyst loadings (0.0019–0.0075 mol%) using Zr complexes 35a and 35b with [OSSO]-type aryl-substituted bis(phenolate) ligands and modified methylaluminoxane (dMMAO) (Scheme 17) [57]. The catalytic system predominantly produced dimers with terminal vinylidene groups (74–91%) and trimers (8–11%) at 25–40 °C for 1 h with the following reagent ratio: [Zr]:[Al]:[1-alkene] = 1:(100–300):(13,350–53,500). The TOF values were adjusted by changing the structure of an aryl substituent R1 at the ortho position of a phenolate moiety of the [OSSO] ligand and the number of dMMAO equivalents used. The highest TOF value (up to 11,100 h−1) was observed for phenyl-substituted precatalyst 35a. The authors explained the low alkene conversion (10–77%) in the presence of 35a and 35b with the deactivation of Zr-H active species during oligomerization.
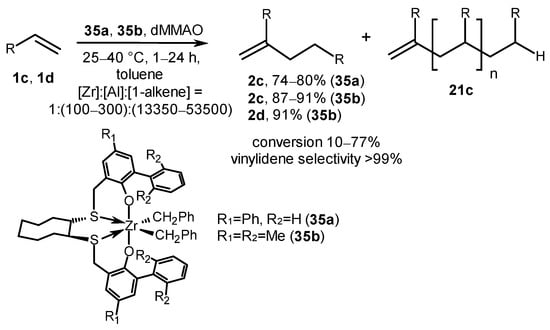
Scheme 17. Post-metallocene Zr complexes of [OSSO] type as catalysts of alkene oligomerization [57].
Bis-phenolate titanium complexes 36a–c, activated by B(C6F5)3 ([Ti]/[B] = 1), catalyzed the transformation of 1-hexene into vinylene oligomers with high yield (up to 97%) and selectivity (99%) (Scheme 18) [58]. Zirconium (36d) and hafnium analogs (36e) showed significantly lower activity (yield of up to 22%), but better selectivity towards vinylidene oligomers (up to 95%). This dependence of regioselectivity on the nature of the transition metal was confirmed in an experiment with 13C-labeled hexene: the cross-linking of an alkene in the case of a Zr catalyst occurs as successive stages of a 1,2-insertion of an olefin into M-H species, a 1,2-insertion of an alkene into M-Alkyl, and β-H elimination. In the case of Ti, the stages of 1,2-olefin insertion into M-H, 2,1-alkene insertion into M-Alkyl, and β-H elimination occur. The rate of 2,1-olefin insertion is affected by solvation, an increase in the bulkiness of the ligand and the growing chain, as well as temperature. Thus, low temperatures down to −80 °C in the case of 36a led to the following ratio: [vinylene]/[vinylidene] = 52/48.
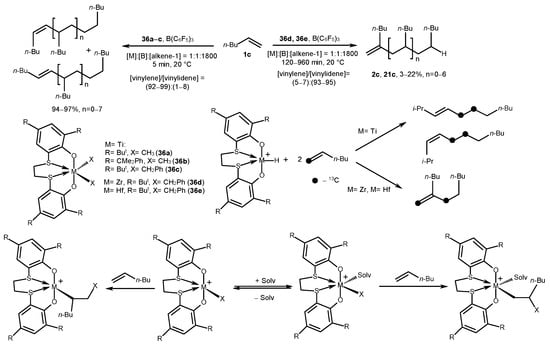
Scheme 18. Post-metallocene bis-phenolate Zr complexes of [OSSO] type as catalysts of alkene oligomerization [58].
Dimers and oligomers of terminal alkenes were synthesized in catalytic systems based on various zirconocenes (3 and 37–52) with cyclopentadienyl ligands; indenyl ligands; fluorenyl ligands, including ansa-complexes; and heterocenes, which were activated in several steps by AlBui3, Et2AlCl, and methylaluminoxane (Scheme 19) [5][40][59][60][61][62]. Cyclopentadienyl complexes, including Cp2ZrCl2 (3), (Me2C)2Cp2ZrCl2 (37), (Me2Si)2Cp2ZrCl2 (38), and OSiMe2Cp2ZrCl2 (39), at low AlMAO/Zr ratios (1–10), catalyzed the regio- and chemoselective formation of head-to-tail α-olefin dimers with yields of 82–94% and 100% alkene conversion [40][61]. The oligomers of α-olefins (1-hexene, 1-octene, and 1-decene) were obtained in the reactions, catalyzed by zirconocenes 40, 41, 42, and 48 and organoaluminum cocatalysts at the following ratio: [Zr]:[AlBui3]:[MAO]:[1-alkene] = 1:20:10:2000 [40][59][60][62]. A yield of 1-hexene dimer decreased to 40–52% and a yield of oligomers increased to 55–57% under the same conditions in the presence of the CpIndZr2Cl2 complex (44) [59][60]. Higher 1-hexene oligomers with Mw = 3900 Da were produced by the Ind2ZrCl2 complex (45) [59]. The TOF values were (1–2.4)·105 h−1 when 46 and 47 were used in the oligomerization of alkenes 1c, 1d, 1u, and 1x [40].
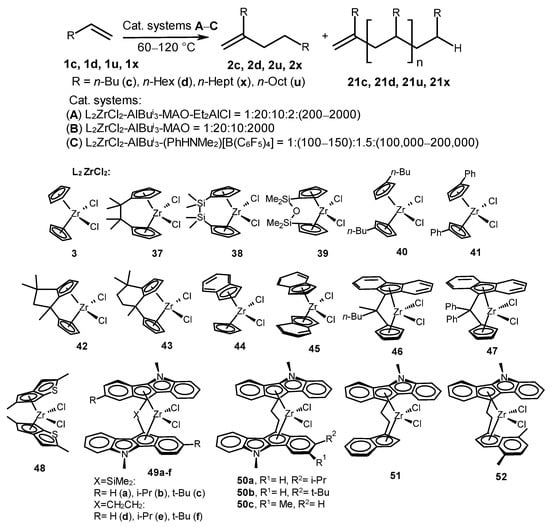
Scheme 19. Alkene dimerization and oligomerization catalyzed by complexes 3 and 37–52.
To explain the catalytic action of the systems, the mechanisms presented in Scheme 20, Scheme 21 and Scheme 22 were suggested. For example, Zr,Al complex A stabilized by the ClMAO– anion [63] formed in the reaction of Cp2ZrCl2 with AlBui3 and MAO was proposed as a catalytically active center for the alkene dimerization reaction (Scheme 20) [59]. An excess of OAC (MAO or AlBui3) increases the amount of dihydride complex B. Catalytically active center A coordinates an alkene molecule at the Zr–H bond to form alkyl complex A1, the further alkylation of which provides intermediate A2. Chlorine atoms in complex A2 promote the process of β-H transfer to the Zr atom, rather than the coordination of the third and subsequent substrate molecules (chain growth). An alkene dimer and catalytically active center A are formed after β-H transfer to a Zr atom. Intermediate B is electrophilic and seems to be sterically accessible for α-olefin oligomerization. An increase in selectivity of the reaction observed in the dimerization after the treatment of a reaction mixture with Et2AlCl is probably due to the conversion of B to A (Scheme 20). The selectivity of the dimerization reaction of α-olefins, therefore, depends mainly on the ratio of catalytically active sites A and B.
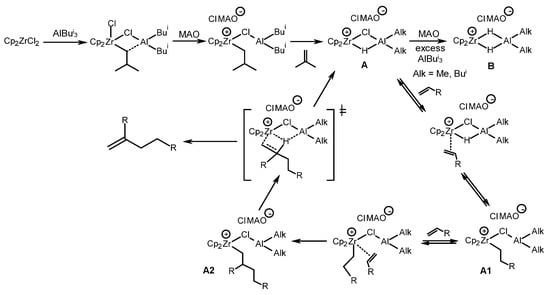
Scheme 20. α-Olefin dimerization mechanism [59].
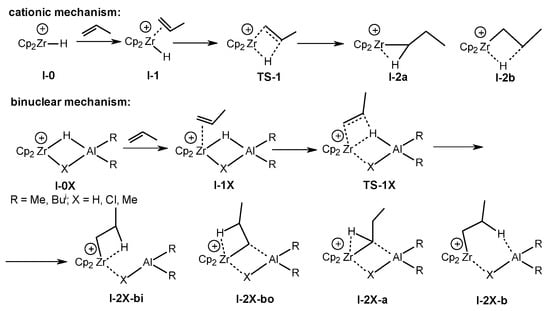
Scheme 21. DFT modeling of the initiation stages of propene dimerization and oligomerization for cationic and binuclear mechanisms [62].
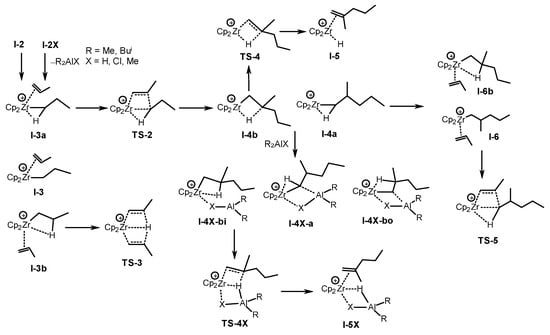
Scheme 22. DFT modeling of propagation and termination stages of the propene dimerization and oligomerization [62].
The initial stages of the propene dimerization and oligomerization with the participation of Zr,Al–complexes were simulated at the DFT M-06X/DGDZVP level of theory to confirm the proposed mechanism [62]. The profiles of propene oligomerization reactions catalyzed by [Cp2ZrH]+ cation I-0 and cationic bimetallic complexes [Cp2Zr(µ-H)(µ-X)AlR2]+ (X = H, Cl, and Me; R = Me and Bui) I-0X were constructed (Scheme 21). Further, activation energies were calculated for the two reaction pathways: the formation of a vinylidene propene dimer via TS-4 and the chain growth via TS-5 (Scheme 22).
A difference between the mechanisms for traditional mononuclear [Cp2Zr-alkyl]+ and binuclear [Cp2Zr-alkyl(R2AlX)]+ species is shown (Scheme 22). Without R2AlX coordination, oligomerization is the favored reaction route. When X = H, highly stable β-agostic complexes I-2X-bo form, so the reactions slow down. If X = Cl, the main direction of the action is the formation of vinylidene dimers. The transition states of β-H elimination TS-4X (X = H and Cl) show a Zr-Al concerted effect. If X = Me, then there is no significant promotion in the β-H elimination process in TS-4 [62].
It was found that the use of molecular hydrogen at a low MAO concentration leads to the results being not typical for Ziegler–Natta processes [62]. The dimerization accelerates, and the selectivity of the reaction in this pathway increases without the formation of hydrogenolysis products in the presence of hydrogen. The DFT simulation showed that the I-2H-bo complex can react with H2 without breaking an H-H bond but with a loss of β-agostic coordination. Molecular hydrogen, therefore, acts as an additional activator for the I-2H-bo hydride complex, which is probably an active and selective catalyst of a dimerization reaction.
Zr heterocene complexes 48 and 49a–f modified with AlBui3 and MMAO-12 were studied in the reaction of 1-decene oligomerization in molecular hydrogen at a [Zr]:[Al]:[MAO]:[1-alkene] ratio of 1:10:75:50,000 at 80–100 °C (Scheme 13) [64]. The conversion of 1-decene reached 99% in the presence of 49d at 80 °C for 4 h, and the formation of low-viscosity oligomers was observed. As the temperature rises to 100 °C, the content of 1-decene dimer increases to 28%. Nevertheless, heterocene 49f was shown to be the most effective catalyst for the synthesis of low-viscosity 1-decene oligomers among the studied complexes. Moreover, the catalytic system based on complex 49f and an activator, (PhHNMe2)[B(C6F5)4], enabled us to achieve a maximum yield (63 wt%) of the most valuable trimer–tetramer fractions of alkene oligomers at a [Zr]:[Al]:[B]:[1-alkene] ratio of reagents of 1:150:1.5:200,000 in 1 atm H2 at 80–110 °C [64].
Unsymmetrical complexes 50a–c, 51, and 52 in the presence of AlBui3, (PhHNMe2)[B(C6F5)4], and H2 (1 bar) at a [Zr]:[Al]:[B]:[1-alkene] ratio of 1:100:1.5:100,000 at 100 °C catalyzed the formation of light 1-decene oligomers with an alkene conversion rate of 86–99% [65]. A gradual decrease in the reaction temperature as 1-decene was shown to reduce the content of dimers (down to 10%) and increase the proportion of oligomers (up to 84%) in the reaction products.
The authors of [64] presented a mechanism for the activation of zirconocene complexes by isobutylalanes, arylboranes, and MAO, as depicted in Scheme 23. They noticed that the classical mechanism implies the participation of active catalytic species, such as alkyl zirconocene cations (L2Zr-R+ (L2 = η5-ligands)) stabilized by [B(C6F5)4]–, [B(C6F5)3R]–, or XMAO– counterions (X = Cl and Me) (Scheme 23A). The reaction between L2ZrCl2 and AlBui3 produces zirconocene alkyl chloride, L2Zr(Cl)Bui. An excess of AlBui3 or HAlBui2 provides various neutral hydride Zr,Al complexes D and E (Scheme 23B). A cationic hydride bimetallic complex F is generated in the presence of perfluoroarylboranes (Scheme 23B). Under the action of excess ClAlBui2, cation F transforms into dichloride Zr,Al-complex G, which can also be formed by the reaction between L2ZrCl2 and R2Al+. Complex G was isolated and characterized by NMR and an X-ray diffraction analysis [64]. Cationic hydride complex F belongs to the category of dormant states as well as species [L2Zr-(μ-Me)2-AlMe2]+ (H). Alkenyl hydride Zr-(μ-H)-Al complex I formed in the presence of excess AlBui3 is considered to be potentially active towards α-olefins (Scheme 23B) [64]. However, a complex similar to I was shown to be inactive in alkene polymerization [66][67]. Moreover, the reaction mechanism involving metallocenes and AlBui3 should take into account the participation of a cationic species, “AlBui2+”, formed as a result of the reaction of OAC with a boron activator.
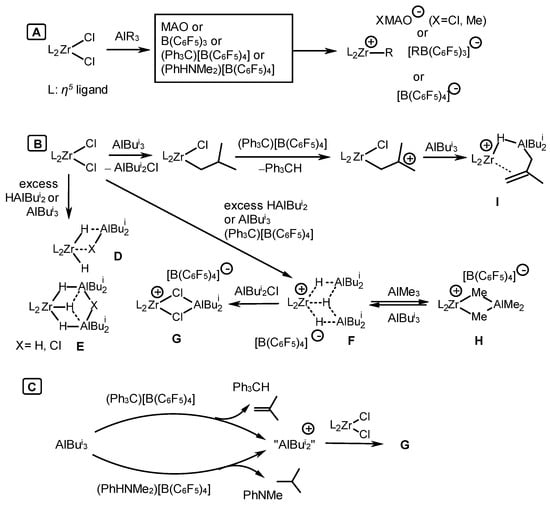
Scheme 23. Catalytic species observed in the systems L2ZrCl2-AlBui3(HAlBui2)-activator [64].
Further, when considering the possible stages of alkene oligomerization (which are coordination, chain growth, and termination, involving the β-H transfer and β-H elimination stages), the authors noted [65] that in the case of heterocenes, the processes of β-H elimination apparently prevail at the final stage of the reaction, when most of the monomer is consumed, leading to the accumulation of C20 dimers in the products (Scheme 24). The β-H elimination can be facilitated by the coordination of R’2AlCl at the Zr center. The competing processes of chain propagation and termination are influenced by both steric and electronic factors of the η5-ligand. It is noted that electron-donating alkyl substituents in the ligand of the complex lead to a decrease in the electrophilicity of the Zr atom and, consequently, to a decrease in catalytic activity, for example, in the case of 49f vs. 50a and 50b. Nevertheless, the lower electrophilicity of Zr (49f) or steric hindrances (for example, in the case of 51 or 52) of the ligand does not promote β-H transfer, which provides higher yields of C30+ or C50+ oligomers [65].
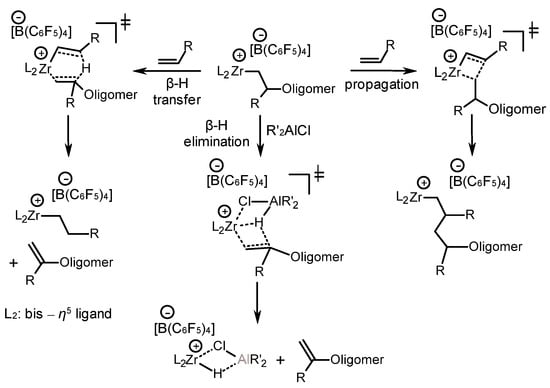
Scheme 24. Various reaction directions in the course of alkene oligomerization [65].
The alkene oligomers were obtained in the reaction catalyzed by ansa-Ph2Si(Cp)(9-Flu)ZrCl2 (53) in the presence of MAO at an Al/Zr of 200 and a temperature of 60 °C (Scheme 25) [68]. This system showed the activity to be 131–155 kg molZr−1 h−1 (PDI = 2.06–2.25). The oligomers constituted a mixture of regioisomeric products with a terminal vinylidene (21) and internal double bonds (54) according to the 1H and 13C NMR spectra. Oligomers with vinylene R′CH=CHR″ and vinylidene CH2=CHR′R″ groups were the major products. Oligomers containing internal disubstituted vinylene groups were formed through 2,1-insertion and β-H elimination or 2,1-insertion and rearrangement, followed by β-H elimination. An NMR analysis of the intensities of the double bond signals and saturated end groups showed the preferential chain transfer to the cocatalyst.
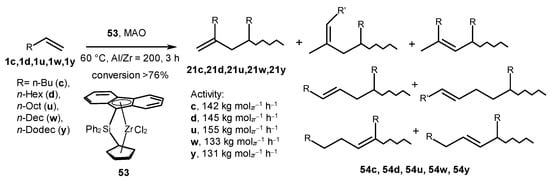
Scheme 25. Alkene oligomers obtained in the reaction catalyzed by ansa-Ph2Si(Cp)(9-Flu)ZrCl2 (53) [68].
Zirconocenes (3, 25, 56–59) and methylaluminoxane catalyzed terminal alkene transformation into dimers 2c, 2d, and 2u with yields of up to 89% at a [Zr]:[MAO]:[1-alkene] reactant ratio of 1:(50–300):(289–2600) and a temperature of 70–100 °C for 3–4 h (Scheme 26) [69]. Catalysts 25 and 59 exhibited the highest activity in the oligomerization reaction. Complex 59 demonstrated superior selectivity towards dimer formation. Dimers 2c, 2d, and 2u were converted into tetramers (55c, 55d, and 55u) under the action of a TiCl4-Et2AlCl system.
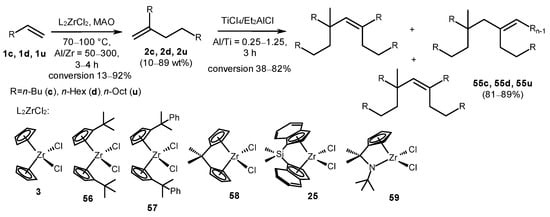
Scheme 26. Alkene transformations into dimers and tetramers [69].
The authors proposed a mechanism for metallocene-catalyzed dimerization based on a structural analysis of alkene dimers (Scheme 27) [69]. The formation of unsaturated (structures A–C) and saturated products (structure D) occurred due to the β-H elimination at cationic metal alkyl centers and chain transfer to a non-transition metal atom (Al), respectively [69]. The vinylidene group (-C=CH2) (structure C) was generated via a 1,2-coordination of an alkene with a [Cp2ZrH]+ cation and the subsequent β-H elimination of the product. Alkene 1,2-coordination, cation rearrangement, and β-H elimination produce structures A and B with trisubstituted vinyl groups (-C=C(CH3)-).
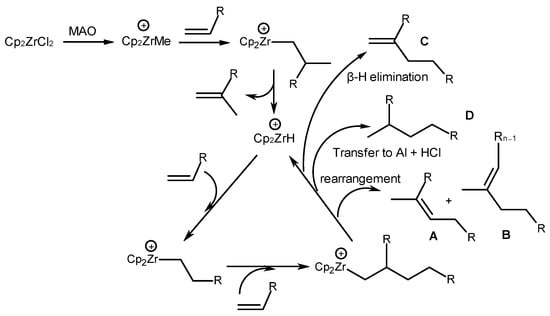
Scheme 27. Mechanism of metallocene-catalyzed dimerization [69].
1-Decene was transformed into oligomers under the action of post-metallocene complexes [M{2,2′-(OC6H2-4,6-tBu2)2NHC2H4NH}(OiPr)2] (60a–c) (M = Ti (a), Zr (b), and Hf (c)) and the (Ph3C)[B(C6F5)4] activator at a [B]:[M] ratio of 1:(0.25–1.5) at 80–120 °C [70] (Scheme 28). The activity of the catalytic system was quantified as 362–484 goligomer mmolcat−1 h−1. The resulting oligomers were characterized by the tacticity values (mm + rr) of 88.5% (Ti), 87.3% (Zr), and 86.8% (Hf); the molecular weight (MN = 445–608 g mol−1); and the PDI value (1.13–1.30). The resulting oligomers differed in structure and contained vinylidene fragments (CH2=CRR′ (21u, δH 4.7–4.8 ppm)), vinyl fragments (CH2=CHR (54u, δH 4.9 and 5.6 ppm)), trisubstituted vinylene groups (RCH=CR′R″ (54u, δH 5.2 ppm)), and disubstituted vinylene groups (RCH=CHR′ (54u; δH 5.3–5.5 ppm)).
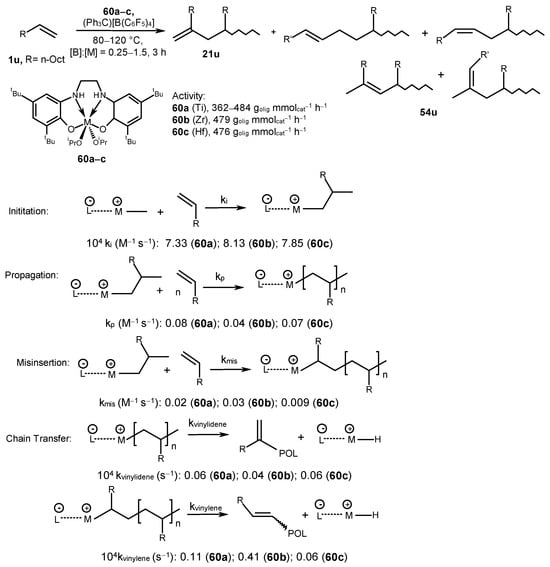
Scheme 28. 1-Decene oligomerization, catalyzed by complexes 60a–c, and kinetic parameters of the reaction [70].
The monomer consumption, the number of active sites, and the number of unsaturated end groups during the oligomerization reaction were evaluated for each catalytic system in the course of the study of the kinetics of 1-decene oligomerization reaction catalyzed by 60a–c (Scheme 28) [70]. An initiation rate constant (ki) in the presence of complex 60b appeared to be higher than those of 60a and 60c (Scheme 28). The ki value was inversely related to the molecular weight of an oligomeric product. A catalyst with a high ki, when the number of active centers is high, leads to low-molecular-weight oligomers. The Ti-based catalytic system exhibited a higher chain propagation rate compared to those of the Zr- and Hf-based systems. Moreover, the reaction initiation stage was found to be slower in comparison to the chain propagation. A decrease in the chain growth constants kp in the Ti > Hf > Zr series was probably due to the electronic nature of metal centers. The rate of formation of a vinylidene product did not depend on the concentration of 1-decene, whereas the rate of formation of a product with an internal double bond was of the first order relative to the monomer concentration. The kvinylidene and kvinylene values were calculated from the initiation rate constants, ki, where kvinylene > kvinylidene by a factor of 2–10. The degree of catalyst involvement in the reaction was 40–60%. The misinsertion stage was slower than the propagation stage for all studied catalysts. The chain termination process runs via the chain β-H transfer to a monomer and the β-H elimination reaction (Scheme 28) [70].
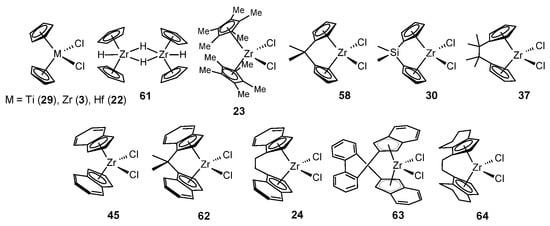
A study of the activity and chemoselectivity of η5-metal complexes 3, 22–24, 29, 30, 37, 45, 58, and 61–64 in the presence of various OACs (HAlBui2, ClAlMe2, ClAlEt2, ClAlBui2, AlMe3, AlEt3, and AlBui3) and activators (MMAO-12, (Ph3C)[B(C6F5)4], and B(C6F5)3) in alkene dimerization and oligomerization showed that either HAlBui2 or AlBui3 at certain ratios ensure the selectivity of the reaction towards dimerization in comparison with AlMe3 or AlEt3 (Scheme 29) [71]. Moreover, Cp2ZrCl2-(AlBui3 or HAlBui2) or [Cp2ZrH2]2-ClAlR2 (R = Me, Et, Bui) systems produced predominantly head-to-tail dimers (2c,d,h,k,u,z) in the presence of MMAO-12 or B(C6F5)3 activators at the [Zr]:[Al]:[MMAO-12]:[1-alkene] ratio of 1:3:30:(50–1000) or the [Zr]:[Al]:[B]:[alkene] ratio of 4:16:1:1000, correspondingly, at 20–60 °C for 5–180 min in toluene with a yield of up to 98% (2c, 98%; 2d, 91%; 2u, 87%; 2z, 95%; 2h, 61%; 2k, 58%) (Scheme 29) [71][72].
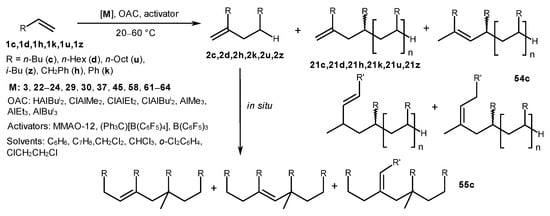
The use of chlorinated solvents (CH2Cl2 and CHCl3) in the Cp2ZrY2-YAlBui2 (Y = H, Cl) systems’ activator (MMAO-12, (Ph3C)[B(C6F5)4]) accelerated the reaction and increased the yield of dimeric products [73]. Under these conditions, the dimers obtained in the first minutes were substrates for the subsequent dimerization and formation of tetramer 55 with yields of up to 79%. Adding an ionic-type cocatalyst, (Ph3C)[B(C6F5)4], to either the Cp2ZrCl2-HAlBui2 or [Cp2ZrH2]2-ClAlBui2 catalytic systems typically resulted in the formation of oligomeric products [72]. Replacing the transition metal atom from Zr to Ti or Hf under the same conditions led to a decrease in activity and selectivity towards dimers [73].
A study on the influence of the ligand structure on the activity and chemoselectivity of the system L2ZrCl2-HAlBui2-MMAO-12 revealed that dimerization occurs with the participation of Zr complexes with sterically unhindered ligands (L = Cp, ansa-Me2CCp2, ansa-(Me2C)2Cp2, and ansa-Me2SiCp2) [74]. Zirconocenes with bulky cyclopentadienyl (L = C5Me5 and rac-H4C2[THInd]2) or electron-withdrawing indenyl (L = Ind, Me2CInd2, H4C2[Ind]2 and BIPh(Ind)2) substituents in the presence of HAlBui2 and MMAO-12 or (Ph3C)[B(C6F5)4] activators predominantly yielded 1-hexene oligomers, which is consistent with the data in Ref. [60]. The assessment of the stereoselectivity of the reaction using 13C NMR spectroscopy showed a dependence of this parameter on the π-ligand environment of the metal and the type of activator [74]. Catalysts with indenyl ligands 45, 62, and 24 were found to be the most stereoselective, demonstrating isotacticity levels of 67%, 93%, and 71%, respectively. An oligomer with an isotacticity level of 67% was obtained under the action of complex 45 in the presence of MMAO-12, whereas (Ph3C)[B(C6F5)4] led to an atactic product. The opposite situation was observed for complex 62 with ansa-bridged ligands: the highest stereoselectivity was achieved in the presence of (Ph3C)[B(C6F5)4].
These facts indicate that a cocatalyst has a significant influence on the stereoregulation process during the alkene coordination through catalytically active centers. As a result, the data on the structure and reactivity of possible intermediates [71][72][74][75], the high selectivity of a reaction towards the dimerization, and completely different rates of oligomerization and dimerization processes allow us to propose a mechanism (Scheme 30). The mechanism implies the involvement of bis-zirconium hydride structures as precursors of dimerization reaction active sites. At the first stage of the reaction, the hydrometalation of alkenes proceeds with the participation of one of the zirconium centers. The introduction of the second alkene molecule, the carbometalation stage, and the β-H elimination stage can also proceed in concert with the involvement of two zirconium atoms. Finally, the dimerization product (2) and the starting bis-zirconium complex are formed. Examples of such bimetallic catalysis are known for the polymerization of alkenes in the presence of subgroup 4 metal complexes [76], as well as ethylene tetramerization reactions on chromium catalysts [77][78][79].
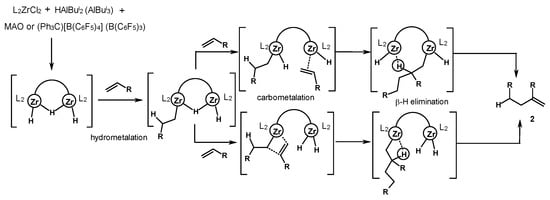
Scheme 30. Probable mechanism of alkene dimerization [72].
Thus, the literature provides extensive information on the dimerization and oligomerization of alkenes under the action of homogeneous catalytic systems based on metallocenes and post-metallocenes. Typically, these works emphasize the key roles of metal hydride intermediates as active species. Therefore, the study of the structure and reactivity of hydride complexes of transition metals is a relevant task to develop models of reaction mechanisms.
References
- Janiak, C. Metallocene and related catalysts for olefin, alkyne and silane dimerization and oligomerization. Coord. Chem. Rev. 2006, 250, 66–94.
- de Klerk, A. Oligomerization. In Fischer-Tropsch Refining; de Klerk, A., Ed.; Wiley-VCH Verlag: Hoboken, NJ, USA, 2011; pp. 369–391.
- McGuinness, D.S. Olefin Oligomerization via Metallacycles: Dimerization, Trimerization, Tetramerization, and Beyond. Chem. Rev. 2011, 111, 2321–2341.
- Osakada, K. (Ed.) Organometallic Reactions and Polymerization; Springer: Berlin/Heidelberg, Germany, 2014; p. 301.
- Nifant’ev, I.; Ivchenko, P.; Tavtorkin, A.; Vinogradov, A.; Vinogradov, A. Non-traditional Ziegler-Natta catalysis in a-olefin transformations: Reaction mechanisms and product design. Pure Appl. Chem. 2017, 89, 1017–1032.
- Nicholas, C.P. Applications of light olefin oligomerization to the production of fuels and chemicals. Appl. Cat. A Gen. 2017, 543, 82–97.
- Busca, G. Acid Catalysts in Industrial Hydrocarbon Chemistry. Chem. Rev. 2007, 107, 5366–5410.
- Lavrenov, A.V.; Karpova, T.R.; Buluchevskii, E.A.; Bogdanets, E.N. Heterogeneous oligomerization of light alkenes: 80 years in oil refining. Catal. Ind. 2016, 8, 316–327.
- Arlman, E.J.; Cossee, P. Ziegler-Natta catalysis III. Stereospecific polymerization of propene with the catalyst system TiCl3*AlEt3. J. Catal. 1964, 3, 99–104.
- Wu, M.M.-S.; Coker, C.L.; Walzer, J.F., Jr.; Jiang, P. Process to Produce Low Viscosity Poly-alpha-Olefins. U.S. Patent 8207390 B2, 26 June 2012.
- Wu, M.M.; Hagemeister, M.P.; Yang, N. Process to Produce Polyalphaolefins. U.S. Patent 8513478 B2, 20 August 2013.
- Comyns, A.E. Encyclopedic Dictionary of Named Processes in Chemical Technology, 4th ed.; CRC Press: Boca Raton, FL, USA, 2014; p. 416.
- Martin, R.W.; Deckman, D.E.; Kelly, K.J.; Emett, C.J.; Hagemeister, M.P.; Harrington, B.A.; Lin, C.-Y.; Matsunaga, P.T.; Ruff, C.J.; Stavens, K.B. Low Viscosity Engine Oil Compositions. U.S. Patent 9234150 B2, 12 January 2016.
- Patil, A.O.; Bodige, S. Synthetic Lubricant Basestocks and Prepared from Vinyl-Terminated Olefin Macromonomers. U.S. Patent 9422497 B2, 23 August 2016.
- Harvey, B.G.; Meylemans, H.A. 1-Hexene: A renewable C6 platform for full-performance jet and diesel fuels. Green Chem. 2014, 16, 770–776.
- Natta, G.; Danusso, F. Stereoregular Polymers and Stereospecific Polymerizations: The Contributions of Giulio Natta and His School to Polymer Chemistry; Symposium Publications Division, Pergamon Press: Long Island City, NY, USA, 1967; p. 888. Available online: https://books.google.ru/books?id=iwQkAQAAMAAJ (accessed on 12 January 2024).
- Fink, G. Polymerization on Molecular Catalysts. In Handbook of Heterogeneous Catalysis; Ertl, G., Helmut Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Fairford, UK, 2008; pp. 3792–3830.
- Nowlin, T.; Mink, R.; Kissin, Y. Supported Magnesium/Titanium-Based Ziegler Catalysts for Production of Polyethylene. In Handbook of Transition Metal Polymerization Catalysts; Hoff, R., Mathers, R.T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 131–155.
- Gardner, B.M.; Seechurn, C.C.C.J.; Colacot, T.J. Industrial Milestones in Organometallic Chemistry. In Organometallic Chemistry in Industry; Colacot, T.J., Seechurn, C.C.C.J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2020; pp. 1–22.
- Kumawat, J.; Gupta, V.K. Fundamental aspects of heterogeneous Ziegler–Natta olefin polymerization catalysis: An experimental and computational overview. Polym. Chem. 2020, 11, 6107–6128.
- Pawlak, M.; Drzeżdżon, J.; Jacewicz, D. The greener side of polymers in the light of d-block metal complexes as precatalysts. Coord. Chem. Rev. 2023, 484, 215122.
- Wilkinson, G.; Birmingham, J.M. Bis-cyclopentadienyl Compounds of Ti, Zr, V, Nb and Ta. J. Am. Chem. Soc. 1954, 76, 4281–4284.
- Long, W.P.; Breslow, D.S. Der Einfluß von Wasser auf die katalytische Aktivität von Bis(π-cyclopentadienyl)titandichlorid-Dimethylaluminiumchlorid zur Polymerisation von Äthylen. Justus Liebigs Ann. Chem. 1975, 3, 463–469.
- Andresen, A.; Cordes, H.-G.; Herwig, J.; Kaminsky, W.; Merck, A.; Mottweiler, R.; Pein, J.; Sinn, H.; Vollmer, H.-J. Halogen-Free Soluble Ziegler Catalysts for the Polymerization of Ethylene. Control of Molecular Weight by Choice of Temperature. Angew. Chem. Int. Ed. 1976, 15, 630–632.
- Bolesłlawski, M.; Pasynkiewicz, S.; Kunicki, A.; Serwatowski, J. Proton magnetic resonance studies on the structure of tetraethylalumoxane. J. Organomet. Chem. 1976, 116, 285–289.
- Yang, X.; Stern, C.L.; Marks, T.J. Cation-like homogeneous olefin polymerization catalysts based upon zirconocene alkyls and tris(pentafluorophenyl)borane. J. Am. Chem. Soc. 1991, 113, 3623–3625.
- Yang, X.; Stern, C.L.; Marks, T.J. Cationic Zirconocene Olefin Polymerization Catalysts Based on the Organo-Lewis Acid Tris(pentafluorophenyl)borane. A Synthetic, Structural, Solution Dynamic, and Polymerization Catalytic Study. J. Am. Chem. Soc. 1994, 116, 10015–10031.
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angew. Chem. Int. Ed. 1995, 34, 1143–1170.
- Chen, E.Y.-X.; Marks, T.J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure−Activity Relationships. Chem. Rev. 2000, 100, 1391–1434.
- Collins, R.A.; Russell, A.F.; Mountford, P. Group 4 metal complexes for homogeneous olefin polymerisation: A short tutorial review. Appl. Petrochem. Res. 2015, 5, 153–171.
- Dzhemilev, U.M.; Ibragimov, A.G. Metal complex catalysis in the synthesis of organoaluminium compounds. Russ. Chem. Rev. 2000, 69, 121–135.
- Guiry, P.J.; Coyne, A.G.; Carroll, A.M. C–E bond formation through hydroboration and hydroalumination. In Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Oxford, UK, 2007; pp. 839–869.
- Dzhemilev, U.M.; Ibragimov, A.G. Hydrometallation of Unsaturated Compounds. In Modern Reduction Methods; Andersson, P.G., Munslow, I.J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 447–489.
- Tolstikov, G.A.; Dzhemilev, U.M.; Tolstikov, A.G. Aluminiyorganicheskie Soedineniya v Organicheskom Sinteze (Organoaluminum Compounds in Organic Synthesis); Akad. Izd. GEO: Novosibirsk, Russia, 2009; p. 645. ISBN 978-645-9747-0147-9744.
- Zaidlewicz, M.; Wolan, A.; Budny, M.M. 8.24 Hydrometallation of C=C and C=C bonds. Group 3. In Comprehensive Organic Synthesis II; Knöchel, P., Gary, A., Eds.; Elsevier Science & Technology Books: Amsterdam, The Netherlands, 2014; Volume 877–963.
- Skupinska, J. Oligomerization of alpha-olefins to higher oligomers. Chem. Rev. 1991, 91, 613–648.
- Janiak, C.; Blank, F. Metallocene Catalysts for Olefin Oligomerization. Macromol. Symp. 2006, 236, 14–22.
- Belov, G.P. Selective dimerization, oligomerization, homopolymerization and copolymerization of olefins with complex organometallic catalysts. Russ. J. Appl. Chem. 2008, 81, 1655–1666.
- Breuil, P.-A.R.; Magna, L.; Olivier-Bourbigou, H. Role of Homogeneous Catalysis in Oligomerization of Olefins: Focus on Selected Examples Based on Group 4 to Group 10 Transition Metal Complexes. Catal. Lett. 2015, 145, 173–192.
- Nifant’ev, I.; Ivchenko, P. Fair Look at Coordination Oligomerization of Higher α-Olefins. Polymers 2020, 12, 1082.
- Olivier-Bourbigou, H.; Breuil, P.A.R.; Magna, L.; Michel, T.; Fernandez Espada Pastor, M.; Delcroix, D. Nickel Catalyzed Olefin Oligomerization and Dimerization. Chem. Rev. 2020, 120, 7919–7983.
- Patel, N.; Valodkar, V.; Tembe, G. Recent developments in catalyst systems for selective oligomerization and polymerization of higher α-olefins. Polym. Chem. 2023, 14, 2542–2571.
- Slaugh, L.H.; Schoenthal, G.W. Vinylidene Olefin Process. U.S. Patent 4658078, 14 April 1987.
- Kondakov, D.Y.; Negishi, E.-I. Zirconium-catalyzed enantioselective methylalumination of monosubstituted alkenes. J. Am. Chem. Soc. 1995, 117, 10771–10772.
- Christoffers, J.; Bergman, R.G. Catalytic Dimerization Reactions of α-Olefins and α,ω-Dienes with Cp2ZrCl2/Poly(methylalumoxane): Formation of Dimers, Carbocycles, and Oligomers. J. Am. Chem. Soc. 1996, 118, 4715–4716.
- Christoffers, J.; Bergman, R.G. Zirconocene-alumoxane (1:1)—A catalyst for the selective dimerization of α-olefins. Inorg. Chim. Acta 1998, 270, 20–27.
- Kretschmer, W.P.; Troyanov, S.I.; Meetsma, A.; Hessen, B.; Teuben, J.H. Regioselective Homo- and Codimerization of α-Olefins Catalyzed by Bis(2,4,7-trimethylindenyl)yttrium Hydride. Organometallics 1998, 17, 284–286.
- Wahner, U.M.; Brüll, R.; Pasch, H.; Raubenheimer, H.G.; Sanderson, R.D. Oligomerisation of 1-pentene with metallocene catalysts. Angew. Makromol. Chem. 1999, 270, 49–55.
- Brüll, R.; Kgosane, D.; Neveling, A.; Pasch, H.; Raubenheimer, H.; Sanderson, R.; Wahner, U. Synthesis and properties of poly-1-olefins. Macromol. Symp. 2001, 165, 11–18.
- Boccia, A.C.; Costabile, C.; Pragliola, S.; Longo, P. Selective Dimerization of γ-Branched α-Olefins in the Presence of C2v Group-4 Metallocene-Based Catalysts. Macromol. Chem. Phys. 2004, 205, 1320–1326.
- Small, B.L.; Marcucci, A.J. Iron Catalysts for the Head-to-Head Dimerization of α-Olefins and Mechanistic Implications for the Production of Linear α-Olefins. Organometallics 2001, 20, 5738–5744.
- Small, B.L. Tridentate Cobalt Catalysts for Linear Dimerization and Isomerization of α-Olefins. Organometallics 2003, 22, 3178–3183.
- Broene, R.D.; Brookhart, M.; Lamanna, W.M.; Volpe, A.F. Cobalt-Catalyzed Dimerization of α-Olefins to Give Linear α-Olefin Products. J. Am. Chem. Soc. 2005, 127, 17194–17195.
- Hanton, M.J.; Daubney, L.; Lebl, T.; Polas, S.; Smith, D.M.; Willemse, A. Selective dimerisation of α-olefins using tungsten-based initiators. Dalton Trans. 2010, 39, 7025–7037.
- Gunasekara, T.; Preston, A.Z.; Zeng, M.; Abu-Omar, M.M. Highly Regioselective α-Olefin Dimerization Using Zirconium and Hafnium Amine Bis(phenolate) Complexes. Organometallics 2017, 36, 2934–2939.
- Flory, P.J. Molecular Size Distribution in Linear Condensation Polymers1. J. Am. Chem. Soc. 1936, 58, 1877–1885.
- Nakata, N.; Nakamura, K.; Ishii, A. Highly Efficient and 1,2-Regioselective Method for the Oligomerization of 1-Hexene Promoted by Zirconium Precatalysts with -Type Bis(phenolate) Ligands. Organometallics 2018, 37, 2640–2644.
- Lian, B.; Beckerle, K.; Spaniol, T.P.; Okuda, J. Regioselective 1-Hexene Oligomerization Using Cationic Bis(phenolato) Group 4 Metal Catalysts: Switch from 1,2- to 2,1-Insertion. Angew. Chem. Int. Ed. 2007, 46, 8507–8510.
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Ivchenko, P.V. Zirconocene-catalyzed dimerization of 1-hexene: Two-stage activation and structure–catalytic performance relationship. Catal. Commun. 2016, 79, 6–10.
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Sedov, I.V.; Dorokhov, V.G.; Lyadov, A.S.; Ivchenko, P.V. Structurally uniform 1-hexene, 1-octene, and 1-decene oligomers: Zirconocene/MAO-catalyzed preparation, characterization, and prospects of their use as low-viscosity low-temperature oil base stocks. Appl. Catal. A Gen. 2018, 549, 40–50.
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Churakov, A.V.; Ivchenko, P.V. Synthesis of zirconium(III) complex by reduction of O2ZrCl2and its selectivity in catalytic dimerization of hex-1-ene. Mendeleev Commun. 2018, 28, 467–469.
- Nifant’ev, I.; Vinogradov, A.; Vinogradov, A.; Karchevsky, S.; Ivchenko, P. Zirconocene-Catalyzed Dimerization of α-Olefins: DFT Modeling of the Zr-Al Binuclear Reaction Mechanism. Molecules 2019, 24, 3565.
- Kuklin, M.S.; Hirvi, J.T.; Bochmann, M.; Linnolahti, M. Toward Controlling the Metallocene/Methylaluminoxane-Catalyzed Olefin Polymerization Process by a Computational Approach. Organometallics 2015, 34, 3586–3597.
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Bagrov, V.V.; Churakov, A.V.; Minyaev, M.E.; Kiselev, A.V.; Salakhov, I.I.; Ivchenko, P.V. A competetive way to low-viscosity PAO base stocks via heterocene-catalyzed oligomerization of dec-1-ene. Mol. Catal. 2022, 529, 112542.
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Bagrov, V.V.; Kiselev, A.V.; Minyaev, M.E.; Samurganova, T.I.; Ivchenko, P.V. Heterocene Catalysts and Reaction Temperature Gradient in Dec-1-ene Oligomerization for the Production of Low Viscosity PAO Base Stocks. Ind. Eng. Chem. Res. 2023, 62, 6347–6353.
- Bryliakov, K.P.; Talsi, E.P.; Semikolenova, N.V.; Zakharov, V.A.; Brand, J.; Alonso-Moreno, C.; Bochmann, M. Formation and structures of cationic zirconium complexes in ternary systems rac-(SBI)ZrX2/AlBu3i/ (X=Cl, Me). J. Organomet. Chem. 2007, 692, 859–868.
- Götz, C.; Rau, A.; Luft, G. Ternary metallocene catalyst systems based on metallocene dichlorides and AlBu3i/: NMR investigations of the influence of Al/Zr ratios on alkylation and on formation of the precursor of the active metallocene species. J. Mol. Catal. A Chem. 2002, 184, 95–110.
- Shao, H.; Wang, R.; Li, H.; Guo, X.; Jiang, T. Synthesis of low-molecular-weight poly-α-olefins using silicon-bridged zirconocene catalyst for lubricant basestock. Arab. J. Chem. 2018, 13, 2715–2721.
- Dong, S.Q.; Mi, P.K.; Xu, S.; Zhang, J.; Zhao, R.D. Preparation and Characterization of Single-Component Poly-α-olefin Oil Base Stocks. Energy Fuels 2019, 33, 9796–9804.
- Hanifpour, A.; Bahri-Laleh, N.; Nekoomanesh-Haghighi, M.; Poater, A. Group IV diamine bis(phenolate) catalysts for 1-decene oligomerization. Mol. Catal. 2020, 493, 111047.
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K. Bimetallic Zr, Zr-Hydride Complexes in Zirconocene Catalyzed Alkene Dimerization. Molecules 2020, 25, 2216.
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R. Catalytic Systems Based on Cp2ZrX2 (X = Cl, H), Organoaluminum Compounds and Perfluorophenylboranes: Role of Zr, Zr- and Zr, Al-Hydride Intermediates in Alkene Dimerization and Oligomerization. Catalysts 2021, 11, 39.
- Kovyazin, P.V.; Bikmeeva, A.K.; Islamov, D.N.; Yanybin, V.M.; Tyumkina, T.V.; Parfenova, L.V. Ti Group Metallocene-Catalyzed Synthesis of 1-Hexene Dimers and Tetramers. Molecules 2021, 26, 2775.
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R.; Ivchenko, P.V.; Nifant’ev, I.E.; Khalilov, L.M. Catalytic Properties of Zirconocene-Based Systems in 1-Hexene Oligomerization and Structure of Metal Hydride Reaction Centers. Molecules 2023, 28, 2420.
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R.; Ivchenko, P.V.; Nifant’ev, I.E. Activation of metallocene hydride intermediates by methylaluminoxane in alkene dimerization and oligomerization. React. Kinet. Mech. Catal. 2023.
- McInnis, J.P.; Delferro, M.; Marks, T.J. Multinuclear Group 4 Catalysis: Olefin Polymerization Pathways Modified by Strong Metal-Metal Cooperative Effects. Acc. Chem. Res. 2014, 47, 2545–2557.
- Rebenstorf, B.; Larsson, R. Why do homogeneous analogs of phillips (CrO3/SiO2) and union carbide (Chromocene/SiO2) polyethylene catalysts fail? Some answers from ir investigations. J. Mol. Catal. 1981, 11, 247–256.
- Brückner, A.; Jabor, J.K.; McConnell, A.E.C.; Webb, P.B. Monitoring Structure and Valence State of Chromium Sites during Catalyst Formation and Ethylene Oligomerization by in Situ EPR Spectroscopy. Organometallics 2008, 27, 3849–3856.
- Rosenthal, U.; Müller, B.H.; Peulecke, N.; Peitz, S.; Wöhl, A.; Müller, W.; Olivier-Bourbigou, H.; Magna, L.; van Leeuwen, P.W.N.M.; Tschan, M.J.L.; et al. Oligomerization, Cyclooligomerization, Dimerization. In Applied Homogeneous Catalysis with Organometallic Compounds; Cornils, B., Herrmann, W.A., Beller, M., Paciello, R., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 307–410.
More
Information
Subjects:
Chemistry, Applied
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
720
Revisions:
2 times
(View History)
Update Date:
24 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No