Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vincenzo Montinaro | -- | 2298 | 2024-01-22 19:39:58 | | | |
| 2 | Catherine Yang | -2 word(s) | 2296 | 2024-01-23 01:43:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Covella, B.; Giliberti, M.; Montinaro, A.; Rossi, L.; Montinaro, V. Hereditary Angioedema (HAE). Encyclopedia. Available online: https://encyclopedia.pub/entry/54213 (accessed on 19 July 2026).
Covella B, Giliberti M, Montinaro A, Rossi L, Montinaro V. Hereditary Angioedema (HAE). Encyclopedia. Available at: https://encyclopedia.pub/entry/54213. Accessed July 19, 2026.
Covella, Bianca, Marica Giliberti, Adriano Montinaro, Luigi Rossi, Vincenzo Montinaro. "Hereditary Angioedema (HAE)" Encyclopedia, https://encyclopedia.pub/entry/54213 (accessed July 19, 2026).
Covella, B., Giliberti, M., Montinaro, A., Rossi, L., & Montinaro, V. (2024, January 22). Hereditary Angioedema (HAE). In Encyclopedia. https://encyclopedia.pub/entry/54213
Covella, Bianca, et al. "Hereditary Angioedema (HAE)." Encyclopedia. Web. 22 January, 2024.
Copy Citation
Hereditary angioedema (HAE) is a rare disease caused by a genetic alteration of the SERPING1 gene and characterized by recurrent attacks of angioedema that involve the skin, and the mucosae of the gastrointestinal tract and upper airways, which significantly affect the quality of life of patients.
angioedema
hereditary
C1 inhibitor
pharmacological
treatment
bradykinin
1. Introduction
Hereditary angioedema (HAE) is a clinical condition characterized by recurrent episodes of swelling that may affect the subcutaneous tissues of the extremities, trunk, face, or upper airways or the gastrointestinal mucosae or genitalia. It is a rare condition that affects 1:50,000 to 1:100,000 people in the general population and is determined by mutations of a specific gene called SERPING1, which codifies for a protein called C1 inhibitor, a regulatory component of the classical complement pathway that also shows anti-enzymatic activity that regulates the activation of several proteases included in the coagulation, fibrinolytic, and contact system pathways [1]. It is now clear that the specific pathogenesis of angioedema attacks resides in a lack of the regulatory activity of C1 inhibitor on the contact system enzymes pre-kallikrein and Factor XII, which normally undergo a mutual activation after modifications that occur on the endothelial surface [2][3]. Kallikrein acts on high molecular weight kininogen and generates a nonapeptide called bradykinin. This latter vasoactive peptide binds specifically to B2 receptors, G-protein coupled receptors that are constitutively expressed on endothelial cells. B2 receptors induce intracellular transduction mechanisms that, through steric rearrangement and destruction of VE-cadherin, produce an increased permeability of the endothelial layer to plasma fluids and the ultimate translocation of liquid from the intravascular space to the subcutaneous site of the derma. HAE patients do not show an increased risk of bleeding or thrombosis [4][5].
HAE is a genetic disease with an autosomal dominant pattern of transmission, and almost all patients show a heterozygous state, with one functioning allele. Homozygous HAE patients have been described but are very rare [6]. Typically, patients will have reduced circulating antigenic (type I) or functional (type II) levels of C1 inhibitor [7]. Due to the pattern of genetic transmission, probands have generally vertical or transversal familial transmission of the disease. However, frequent de novo mutations of the SERPING1 gene (up to 25% of all cases) must be kept in mind for those cases that do not show a familial pattern. Mutations of the SERPING1 gene, especially those that produce a type I phenotype (85% of cases), are diverse and include missense, non-sense, frameshift insertion or deletion or slicing defects which are spread all over the gene domains [8]; while mutations that produce a type II phenotype (15% of cases), are generally missense mutations localized in exon 8 [9]; overall, almost 750 different mutations have been reported [10].
2. State of the Art Treatment of Hereditary Angioedema
The objective of treatment of HAE is to reduce the number and severity of attacks and to restitute a normal quality of life to the patients. According to the last WAO/EAACI recommendations these objectives can be reached with approved therapies [11]. However, there are still some unmet needs for which several drugs are undergoing active testing and will probably enrich the therapeutic armamentarium towards the end of this decade.
The mainstay of the actual HAE therapies includes drugs for the treatment of single attacks with an on-demand basis and prophylactic treatments for both short-term pre-procedural prophylaxis (i.e., before surgery or dental procedures) or long-term prophylaxis aimed at preventing HAE attacks and potentially completely abrogating the occurrence of attacks. For on-demand treatment there are currently three drugs approved by the FDA and EMA and one more drug approved only by the FDA [12]. The first three are plasma-derived C1 inhibitor for IV injection (Berinert, CSL Behring, Marburg, Germany), recombinant human C1 inhibitor (Ruconest, Pharming, Leiden, The Netherlands), and icatibant (Firazyr, Takeda Pharmaceutical International AG, Dublin, Ireland), a specific antagonist of bradykinin binding to its receptor B2R, that can be administered by subcutaneous injection. Ecallantide (Kalbitor, Takeda) is the fourth drug approved only by the FDA; this is a polypeptide inhibitor of plasma kallikrein, administered subcutaneously. All these drugs have a different pharmacodynamic profile (Figure 1) and have been proved effective in halting the evolution of HAE attacks, including laryngeal attacks, in RCT studies, thus accelerating the resolution of an attack and providing relief shortly after administration and complete resolution of the attack in a few hours after injection. Moreover, post-registration registries have shown a consistent effect on the repetitive use and safety of these drugs over a long period of time [13].
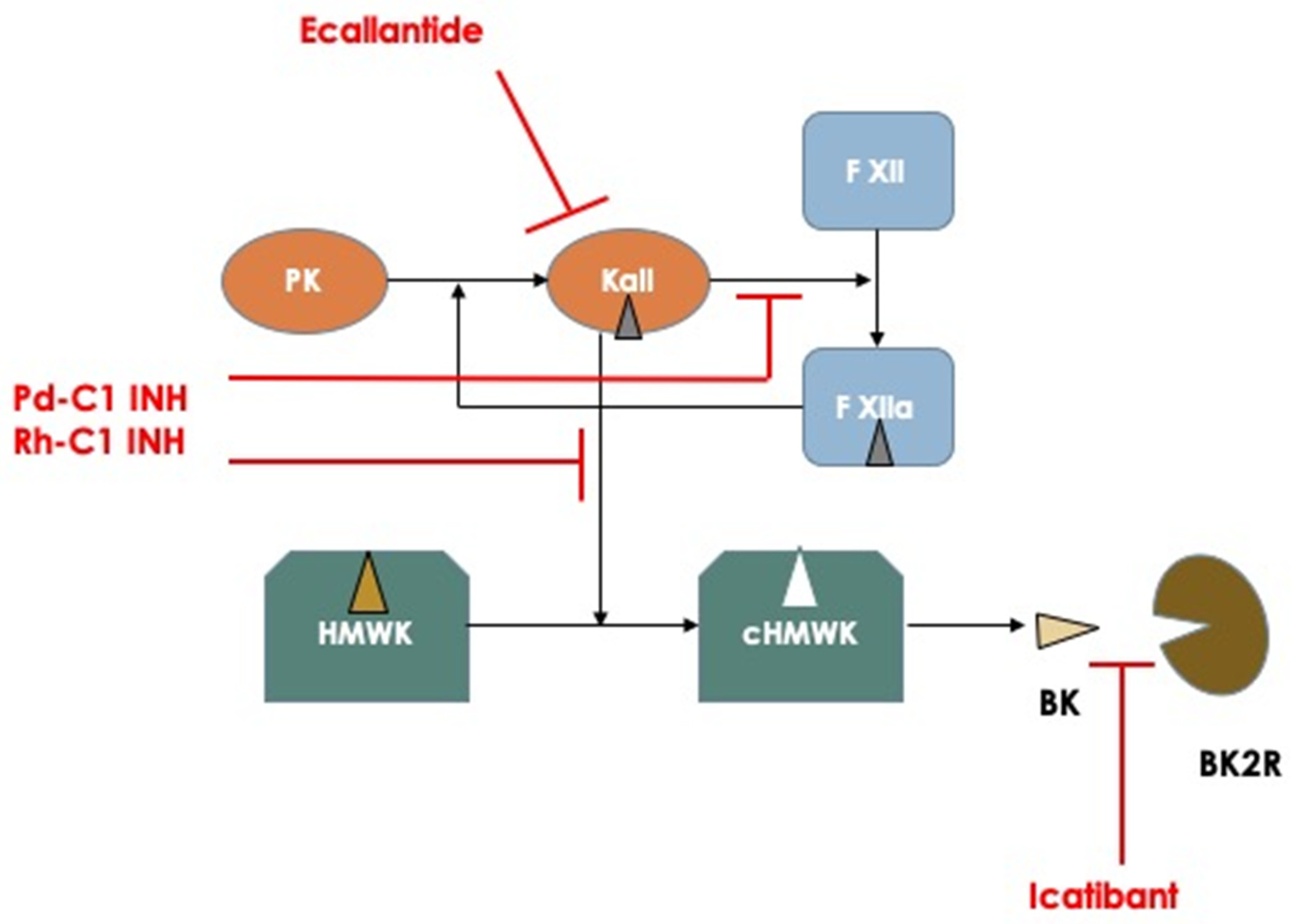
Figure 1. Schematic representation of the principal mechanisms involved in the pathogenesis of hereditary angioedema attacks and pharmacodynamics of the actual available medications used for treating acute attacks. Abbreviations: PK: Prekallikrein; Kall: Kallikrein; F XII: Factor XII; HMWK: High Molecular Weight Kininogen; cHMWK: cleaved HMWK; BK: Bradykinin; BK2R: Bradykinin Receptor type 2.
For short-term prophylaxis, the only approved drug is plasma-derived C1 inhibitor (Bering, CSL Behring; Cinryze, Takeda), which is generally administered intravenously shortly (1 h) before the procedure. Due to the long half-life of plasma-derived C1 inhibitor—up to 32 h—a single injection of the proper dosage is sufficient to provide an adequate coverage even for long surgical procedures. Alternatively, an old approach for short term prophylaxis consisted of the administration of attenuated androgens, like danazol, at high dosage (i.e., 600 mg/day in three doses) for a period of 5 days before the procedure. The effect of danazol in preventing HAE attacks is not completely understood, but probably consists of a combination of a stimulation of C1 inhibitor synthesis and an increase in circulating metalloprotease that normally operate a catabolism of bradykinin. Attenuated androgens are no longer recommended as first line therapies, due to the high rate of adverse effects associated with the use of these drugs.
In the last 10–15 years there has been an increased use of long-term prophylaxis for HAE attacks, especially in patients that present with frequent attacks (>3–4/month) and severe attacks (laryngeal or abdominal). Owing to the availability of novel therapeutic options that include plasma-derived C1 inhibitor and a monoclonal antibody that targets activated plasma kallikrein, the last updated version of the WAO/EAACI guidelines on the management of HAE recommend that the goals of treatment are to achieve total control of the disease and to normalize patient’s lives [11]. The available therapeutic options then include plasma-derived C1 inhibitor for IV infusion (Cinryze, Takeda) or for subcutaneous administration at a higher dosage (Haegarda or Berinert sc, CSL Behring); both types of preparations need a twice-weekly infusion of each drug and can obtain a reduction in the attack rate of between 50% to a maximum of 88% for the subcutaneous C1 inhibitor preparation. Another more recent option for long-term prophylaxis is represented by lanadelumab, a humanized monoclonal antibody against activated plasma kallikrein, which is administered subcutaneously once every two weeks in the induction and once every 28 days after stabilization of the therapeutic response or after 6 months if the patient is attack-free. Lanadelumab can abate the attack rate by up to 73–87% [14]. Finally, an oral inhibitor of activated plasma kallikrein has recently been developed; berotralstat (Orladeyo, BioCryst Pharmaceutical, Dublin, Ireland) can be administered once a day for long-term prophylaxis. It can reduce attack frequency by 45–50% with a tendency toward increased efficacy for longer treatment periods. In Table 1 there is a synthesis of the main characteristics of actually available drugs for the treatment of HAE patients.
Table 1. Approved drugs for the treatment of HAE attacks or short-term or long-term prophylaxis.
| Drug Name (Trademark) | Approved Indications | Dosage | Mechanism of Action |
|---|---|---|---|
| Plasma-derived C1 inhibitor (Berinert, CSL Behring) | Acute attacks STP All age groups |
20 IU/kg iv | Inhibits factor XIIa and activated plasma kallikrein |
| Recombinant human C1 inhibitor (Ruconest, Pharming) | Acute attacks Adolescents and adults |
Inhibits factor XIIa and activated plasma kallikrein | |
| Icatibant (Firazyr, Takeda) * | Acute attacks Adults and age > 2 yr. |
30 mg sc Reduced dosage |
Bradykinin B2-receptor antagonist |
| Ecallantide (Kalbitor, Takeda) | Acute attacks Adults and adolescents |
30 mg sc | Inhibits plasma kallikrein |
| Plasma-derived C1 inhibitor (Cinryze, Takeda) | Acute attacks, STP LTP All age groups |
1000 IU 1000 IU iv every 3–4 days |
Inhibits factor XIIa and activated plasma kallikrein |
| Plasma-derived C1 inhibitor (Haegarda, Berinert sc, CSL Behring) | LTP | 60 IU/kg every 3–4 days | Inhibits factor XIIa and activated plasma kallikrein |
| Lanadelumab (Takhzyro, Takeda) | LTP Adults, adolescents |
300 mg sc every 2 or 4 weeks | Inhibits activated plasma kallikrein |
| Berotralstat (Orladeyo, BioCryst) | LTP Adults, adolescents |
150 mg daily orally | Inhibits activated plasma kallikrein |
* Patent expired in 2019, generic icatibant is now available.
According to the efficacy of available treatments that allow the efficient management of attacks or reduce the frequency of attacks in long-term prophylaxis, but cannot offer complete disease control or normalize patients’ lives, there are some unmet needs that can be met with new drugs. For this reason, several pharmaceutical companies are actively developing new therapeutic options for HAE patients. In the next part, the researchers will review all the novel drugs that are undergoing active testing for efficacy and safety in HAE patients.
3. New Drugs for the Treatment of Hereditary Angioedema
Moving from the actual scenario of the treatment of hereditary angioedema, one can envision some possible unmet needs in the therapy for this specific condition: (1) the possibility to have an effective and rapid drug for treating on demand acute attacks that should allow the avoidance of any type of injective drug; (2) an effective oral drug for long-term prophylaxis with an efficiency of attack abatement comparable to the available injective molecules; (3) an injective drug for long-term prophylaxis with extended efficacy (2–3 months or more); (4) a definitive cure for the disease, possibly by genetic therapy.
Given these unmet needs, the pharmaceutical industry has undertaken a rush to try to fill all these needs. Therefore, several molecules have begun clinical experimentation and are actually under active testing in different phases (Table 2). In Figure 2, all the new tested drugs are shown according to their pharmacodynamic target. Several new molecules are undergoing experimentation and they have been developed with different pharmacologic technologies that include: (1) orally active, small inhibiting molecules, (2) monoclonal antibodies, (3) antisense oligonucleotide for RNA translation blockade, and (4) genetic technology for genome editing. According to the targets for which the different drugs have been engineered, it can indicate: (1) activated factor XII, (2) prekallikrein gene, (3) activated plasma kallikrein, (4) Bradykinin B2-receptor, (5) SERPING1 gene (genetic therapy), and (6) prekallikrein gene (genetic therapy) Figure 2.
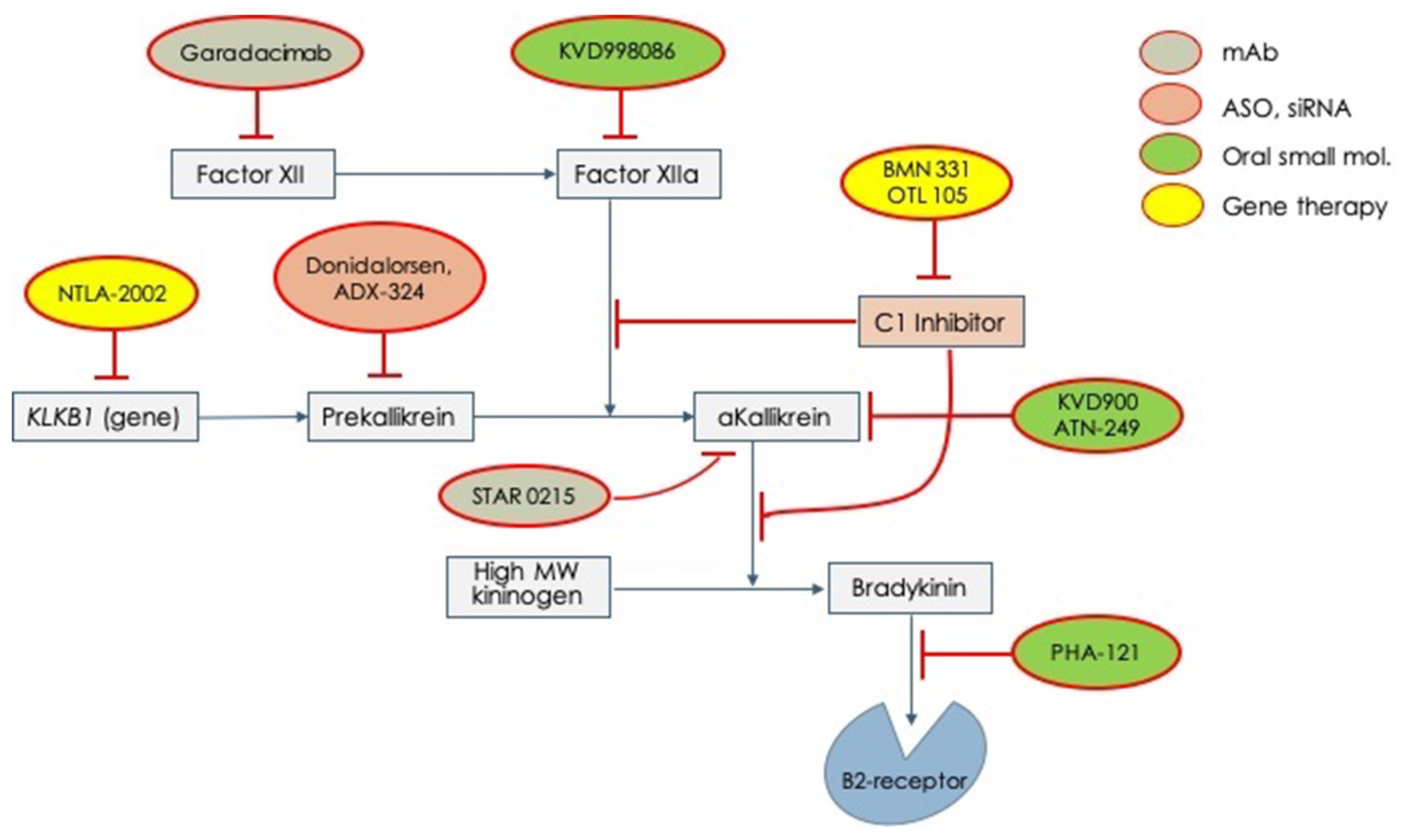
Figure 2. Novel drugs for the treatment of acute attacks or long-term prophylaxis of hereditary angioedema which are undergoing clinical trials for testing efficacy and safety. mAb = monoclonal antibody; ASO = anti-sense oligonucleotide; siRNA = small interfering RNA.
Table 2. Active clinical trials that are testing the efficacy and safety of the novel drugs for the treatment of hereditary angioedema.
| Medication | Producer | Trial Name/ Number |
Therapy | Pharmacodynamics | Route | Phase | Estimated Completion |
|---|---|---|---|---|---|---|---|
| KVD900 | KalVista Pharmaceuticals, Ltd. | KONFIDENT NCT05259917 |
ODT | Inhibitor of plasma Kallikrein | Oral | 3 | October 2023 |
| Deucrictibant PHA-121 | Pharvaris | RAPIDe-2 NCT05396105 |
ODT | Inhibitor of B2-bradykinin receptor | Oral | 2/3 | December 2024 |
| Deucrictibant PHA-121 | Pharvaris | HAE-CHAPTER-1 NCT05047185 |
LTP | Inhibitor of B2-bradykinin receptor | Oral | 2 | December 2026 |
| Garadacimab | CSL Behring | NCT04739059 | LTP | mAb, Inhibitor of activated Factor XII | Subcutaneous | 3b | November 2025 |
| STAR-0215 | Astria Therapeutics Inc. | ALPHA-STAR NVT05695248 |
LTP | mAb, inhibitor plasma Kallikrein | Subcutaneous | 1b/2 | November 2024 |
| STAR-0215 | Astria Therapeutics Inc. | ALPHA-SOLAR NCT06007677 |
LTP | mAb, inhibitor plasma Kallikrein | Subcutaneous 1 dose every 3 or 6 months |
2 | October 2029 |
| Donidalorsen IONIS-PKK-LRx |
Ionis Pharmaceuticals, Inc. | OASIS-HAE NCT05139810 |
LTP | ASO, inhibitor plasma Kallikrein | Subcutaneous 1 dose every 4 or 8 weeks |
3 | April 2024 |
| ADX-324 | ADARx Pharmaceuticals | NCT05691361 | LTP | siRNA, inhibitor of PKK synthesis | Subcutaneous | 1 | November 2024 |
| NTLA-2002 | Intellia Therapeutics | NCT05120830 | Curation | CRISP-Cas9 gene editing, KLKB1 gene | Intravenous | 1/2 | December 2025 |
| BMN 331 | BioMarin Pharmaceutical | HAERMONY-1 NCT05121376 |
Curation | AAV5-vector SERPING1 gene therapy | Intravenous | 1/2 | November 2028 |
4. Genetic Therapies for the Curation of Hereditary Angioedema
The ultimate objective of any novel therapy for treating hereditary angioedema is curation of the disease. This can be obtained by manipulating the genome by inserting a new SERPING1 gene that will compensate for the mutated allele and, therefore, will restitute a condition similar to normality with two alleles producing a normal amount of the C1 inhibitor protein and, at the bottom, abrogating the predisposing condition for recurrence of angioedema attacks. The other modality of genetic therapy that is being explored for curation involves the manipulation of a different target gene, without the insertion of a new SERPING1 gene. A gene target is KLKB1, which codifies for plasma prekallikrein, the precursor of activated plasma kallikrein, a central enzyme in the generation of the ultimate mediator of angioedema attacks: bradykinin.
BioMarin pharmaceutical (San Raphael, CA, USA) is developing an adenovirus-based vector to carry the wild-type SERPING1 gene within the genome of HAE patients, which could determine a curation of the disease with one-time gene therapy. The specific product BMN 331 is under experimentation [HAErmony-1] with a phase 1–2 single arm, dose escalation, open label trial that is testing AAV5 hSERPING1 in patients with HAE type I or II [NCT05121376]; the gene sequence is under the control of a liver-specific promoter, so that the specific protein can be produced and released only by hepatocytes. Patients will receive a single IV injection of the BMN 331 at a dose that is selected from three doses and will be followed-up for 5 years thereafter, monitoring efficacy (levels of plasma C1 inhibitor) and safety. Completion of the study is estimated for November 2028, and at the moment no information has been released by the manufacturer.
The alternative approach is to use CRISP-Cas9 technology to perform gene editing of the KLKB1 gene, that codifies for prekallikrein. NTLA-2002 (Intellia Therapeutics, Inc., Cambridge, MA, USA) contains genetic material that, once included in the genome, will deactivate the KLKB1 gene, producing a significant reduction of circulating plasma levels of prekallikrein. Some interim data from the phase 1 and 2 trial [NCT05120830] were presented at the American College of Allergy, Asthma and Immunology in November 2022. They included data from 10 enrolled patients that had received 25, 50, or 75 mg IV injections of NTLA-2002. After 32 weeks, patients presented a reduction of plasma prekallikrein by 64–92% according to different dosages. Patients showed an attack-free period lasting up to almost 1 year, with a global reduction in the attack rate by 95%. Adverse effects were only mild, and the treatment showed a good safety profile [15].
References
- Longhurst, H.; Cicardi, M. Hereditary angio-oedema. Lancet 2012, 379, 474–481.
- Reshef, A.; Kidon, M.; Leibovich, I. The Story of Angioedema: From Quincke to Bradykinin. Clin. Rev. Allergy Immunol. 2016, 51, 121–139.
- Zuraw, B.L.; Christiansen, S.C. HAE Pathophysiology and Underlying Mechanisms. Clin. Rev. Allergy Immunol. 2016, 51, 216–229.
- Joseph, K.; Tholanikunnel, T.E.; Kaplan, A.P. Treatment of episodes of hereditary angioedema with C1 inhibitor: Serial assessment of observed abnormalities of the plasma bradykinin-forming pathway and fibrinolysis. Ann. Allergy Asthma Immunol. 2010, 104, 50–54.
- Reshef, A.; Zanichelli, A.; Longhurst, H.; Relan, A.; Hack, C.E. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy 2015, 70, 506–513.
- Bafunno, V.; Divella, C.; Sessa, F.; Tiscia, G.L.; Castellano, G.; Gesualdo, L.; Margaglione, M.; Montinaro, V. De novo homozygous mutation of the C1 inhibitor gene in a patient with hereditary angioedema. J. Allergy Clin. Immunol. 2013, 132, 748–750.e3.
- Germenis, A.E.; Speletas, M. Genetics of Hereditary Angioedema Revisited. Clin. Rev. Allergy Immunol. 2016, 51, 170–182.
- Pappalardo, E.; Caccia, S.; Suffritti, C.; Tordai, A.; Zingale, L.C.; Cicardi, M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: Functional and structural correlates. Mol. Immunol. 2008, 45, 3536–3544.
- Zahedi, R.; Aulak, K.S.; Eldering, E.; Davis, A.E., 3rd. Characterization of C1 inhibitor-Ta. A dysfunctional C1INH with deletion of lysine 251. J. Biol. Chem. 1996, 271, 24307–24312.
- Santacroce, R.; D’Andrea, G.; Maffione, A.B.; Margaglione, M.; d’Apolito, M. The Genetics of Hereditary Angioedema: A Review. J. Clin. Med. 2021, 10, 2023.
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990.
- Christiansen, S.C.; Zuraw, B.L. Hereditary angioedema: On-demand treatment of angioedema attacks. Allergy Asthma Proc. 2020, 41, S26–S29.
- Maurer, M.; Aberer, W.; Caballero, T.; Bouillet, L.; Grumach, A.S.; Botha, J.; Andresen, I.; Longhurst, H.J.; IOS Study Group. The Icatibant Outcome Survey: 10 years of experience with icatibant for patients with hereditary angioedema. Clin. Exp. Allergy 2022, 52, 1048–1058.
- Zanichelli, A.; Montinaro, V.; Triggiani, M.; Arcoleo, F.; Visigalli, D.; Cancian, M. Emerging drugs for the treatment of hereditary angioedema due to C1-inhibitor deficiency. Expert. Opin. Emerg. Drugs 2022, 27, 103–110.
- Longhurst, H.; Fijen, L.M.; Lindsay, K.; Butler, J.; Golden, A.; Maag, D.; Xu, Y.; Cohn, D.M. In vivo CRISP/Cas 9 editing of KLKB1 in patients with hereditary angioedema: A first in-human study. In Proceedings of the ACAAI Annual Scientific Meeting 2022, Louisville, KY, USA, 10–14 November 2022.
More
Information
Subjects:
Medicine, General & Internal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
626
Revisions:
2 times
(View History)
Update Date:
23 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No