 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tatiana Armeni | -- | 2946 | 2024-01-22 15:32:43 | | | |
| 2 | Lindsay Dong | Meta information modification | 2946 | 2024-01-23 02:59:04 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2946 | 2024-01-23 03:00:11 | | | | |
| 4 | Lindsay Dong | Meta information modification | 2946 | 2024-01-23 03:00:47 | | |
Video Upload Options
Hemoglobin is one of the proteins that are more susceptible to S-glutathionylation and the levels of its modified form, glutathionyl hemoglobin (HbSSG), increase in several human pathological conditions.
1. Introduction
Oxidative stress, caused by an imbalance between oxygen reactive species (ROS) accumulation in cells and the ability of scavenging systems to remove these reactive products, plays a major role in the pathogenesis of a number of diseases. Additionally, the disease severity and progression are linked to the entity which is damaging to various biological structures, namely cellular membranes, lipids, proteins and nucleic acids [1][2]. There are several human diseases where the involvement of oxidative stress has been well documented, including cancer [3][4], diabetes [5], rheumatoid arthritis [6], atherosclerosis [7], cardiovascular disease [8], neurodegenerative disorders such as Parkinson’s, Alzheimer’s and Huntington’s diseases, amyotrophic lateral sclerosis [9][10] and pulmonary, renal, and hepatic diseases [11][12][13]. In the case of neurodegenerative diseases, the initiation or the progression is particularly vulnerable to ROS due to the high metabolic rate, unique lipid composition, and minimal cell turnover of the nervous system. Oxidative stress could also be a causative player in diseases like Alzheimer’s and Parkinson’s disease, amyotrophic lateral sclerosis, and ischemia-reperfusion injuries, considering that copper and iron, able to generate hydroxyl radicals starting from hydrogen peroxide or superoxide, show high levels in specific brain areas [14]. In addition, oxidative stress is well known to be involved in the pathogenesis of certain lifestyle-related disorders, such as hypertension [15], diabetes [16], atherosclerosis [17] and rheumatoid arthritis [18].
Given the strong correlation between oxidative stress and the pathogenesis of several diseases, there is a progressively increasing interest in identifying biomarkers for pathological conditions related to oxidative stress, with the aim of obtaining an accurate evaluation of oxidative stress levels and exploring the efficacy of antioxidant strategies. Among the different systems involved in cell and tissue protection against oxidative damage, glutathione, the main intracellular low-molecular-weight thiol molecule, plays a crucial role. This biological redox agent is able to shield thiol groups of proteins from irreversible oxidation and inactivate ROS, thus reducing and preventing damage caused by free radicals. Glutathione can exist as glutathione (GSH) or glutathione disulfide (GSSG). It can also bind to different proteins through disulfide bond formation, obtaining glutathionylated proteins [19]. Although reversible, this post-translational modification (PTM) has the potential to escalate as a pathological condition, as observed in the S-glutathionylation of actin during prolonged oxidative stress in Friedreich’s ataxia (FRDA). The impact of this continuous modification of actin is to impair its polymerization capacity and its affinity for tropomyosin, compromising the stability of actin filaments and causing a dysregulation of the cytoskeleton [20]. Moreover, in Alzheimer’s disease, S-glutathionylation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) prevents the correct functioning of this protein involved in the glycolytic pathway [21]. Therefore, considering that specific proteins are involved in each disease, PTMs such as S-glutathionylation could contribute to the manifestation of neurodegenerative diseases as well as other disorders associated with oxidative damage [22][23]. Of the several proteins that may undergo S-glutathionylation, hemoglobin has captured the attention of scientists because of both the high susceptibility of red blood cells to oxidative damage and the pivotal role played by glutathione in protecting erythrocytes from oxidative stress. Indeed, oxidative stress to hemoglobin could induce the formation of hemi-chromes and heme degradation products, thus leading to inflammatory processes and tissue damage [24]. To counteract this harmful scenario, glutathione neutralizes ROS using enzymes, like glutathione peroxidase (GPX) and glutathione reductase (GR), and maintains the redox state of hemoglobin, avoiding the generation of damaging methemoglobin [24]. Starting from this evidence, glutathionyl hemoglobin (HbSSG), in which glutathione binds mainly to the cysteine residue Cys93 of β hemoglobin chain by a disulfide bond, has been largely investigated as a protective response against oxidative damage, and its quantification is vital in clinical research because HbSSG reflects the body’s efforts to counteract the impact of ROS.
2. Protein S-Glutathionylation
Glutathione, the main low-molecular-weight cell thiol, is a pivotal antioxidant molecule within mammalian cells, able to contrast oxidizing molecules that alter the cell antioxidant system, such as hydrogen peroxide. Indeed, following the elimination of the oxidizing molecules, GSSG is reconverted into its reduced state (GSH) by GR that uses NADPH as a cofactor [25][26]. The reversible PTM based on the formation of a disulfide bond between the cysteine of glutathione and protein cysteines is called protein S-glutathionylation, and represents one of the S-thiolation forms. This modification is able to communicate changes in the redox state related to GSH and GSSG levels to the cell [25][27]. In particular, S-glutathionylation occurs mostly either by direct oxidation (1) or through the thiol-disulfide exchange process between GSSG and thiol groups of targeted proteins (2) [27][28]. Other mechanisms are possible, because like protein thiols, the cysteine of GSH can be activated through the formation of glutathione sulfenic acid (GS-OH) resulting in protein S-glutathionylation (3) [29][30]. Also, less common mechanisms are described like the thio-sulfinate derivative of GSH (GS(O)SG) or S-nitroso-thiols such as GSNO that could be able to react with protein cysteines or protein thio-sulfinates that could react with GSH [31].
Protein –SH + GSH → Protein-SSG (1)
Protein –SH + GSSG ⇆ Protein-SSG + GSH (2)
Protein –SH + GS-OH → Protein-SSG (3)
In reaction (1) a surface protein sulfhydryl (Protein–SH) becomes activated by ROS through the formation of cysteine sulfenic acid (Protein–S–OH) or cysteine thiyl radical (Protein–S•) and then undergoes S-glutathionylation reacting with GSH. In reaction (2) protein sulfhydryl (Protein –SH) deprotonates to a thiolate anion (Protein–S−) in a basic microenvironment, attacking the sulfhydryl of intracellular GSSG, with the subsequent formation of disulfide (Protein –SSG). In reaction (3), besides the formation of glutathione sulfenic acid (GS-OH), the cysteine of GSH can be activated also in the form of glutathione disulfide S-oxide (GS(O)SG) or S-nitroso-thiols such as GSNO [31]. Under physiological conditions, the role of this reversible PTM is to regulate redox signaling cycle and avoid the irreversible modifications of cysteine residues. Instead, following the onset of severe oxidative stress condition, part of GSH is used to eliminate free radicals, thus leading to a reduction in GSH concentration and an increase of GSSG level. The decrease of GSH/GSSG ratio produces a large spectrum of oxidized biological molecules and overwhelms protein S-glutathionylation (mainly through the above-mentioned thiol-disulfide exchange reaction (2)) trying to prevent the redox signaling disruption [32]. S-glutathionylation can occur spontaneously in a oxidized environment or can be catalyzed by enzymatic systems, such as glutathione S-transferase (GST), peroxiredoxins and sometimes glutaredoxins, able to transfer glutathione molecule to targeted proteins [32]. In particular, among the different cytosolic GST, it has been reported that GSTπ, mainly present in the lung and liver but also in the brain and heart, induces both in vitro and in vivo S-glutathionylation [33][34]. Along with GSTπ, the glyoxalase system and glutaredoxin, according to the ratio between GSH and GSSG, enzymatically promote the creation of glutathione adducts [34][35]. Upon returning to normal redox condition, since it is a reversible modification, S-glutathionylation could be reverted by the thiol-disulfide exchange reverse reaction (deglutathionylation), spontaneously or catalyzed by specific enzymes, called protein thiol-disulfide oxidoreductases [36]. In particular, thioredoxin, glutaredoxin and protein disulfide isomerase, belonging to the protein thiol-disulfide oxidoreductase family, represent pivotal enzymes able to catalyze protein disulfide bond reduction. Given that they can occasionally also induce S-glutathionylation under oxidative stress condition, as reported above, these enzymes contribute to the creation of a highly dynamic redox microenvironment [32]. Thioredoxin is involved in pro-proliferative, anti-apoptotic and anti-oxidative mechanisms, and its glutathionylation causes an impairment of its function [37]. Glutaredoxins, also called thioltransferases, possess the activity of thiol-disulfide exchange, thus reducing glutathionyl proteins. Glutaredoxins have also been identified within erythrocytes, in which they promote the formation of hemoglobin starting from HbSSG [38]. Lastly, protein disulfide isomerase contributes to the reduction, formation and isomerization of protein disulfide bonds, crucial for the correct structure and fold of several proteins, within the mammalian endoplasmic reticulum [39].
Overall, S-glutathionylation is a rapid, specific and reversible reaction, which, in addition to its protective function, can modulate cellular functions, such as metabolism, energy sensing, cell migration and growth, based on the redox state in which the cell is found [40][41]. Protein S-glutathionylation occurs both under physiological conditions and pathological states in which defense mechanisms are triggered against oxidative stress.
3. S-Glutathionylation of Hemoglobin
Among the different proteins that could be glutathonylated there is hemoglobin. Human hemoglobin is a tetrameric protein, composed of two α globin chains and two β globin chains (α2β2), which plays a key role in oxygen transport in red blood cells throughout the body. Indeed, each globin chain contains a heme able to bind one molecule of oxygen. Several intra- and inter-subunit interactions occur between amino acid residues in hemoglobin and are responsible for the dynamic equilibrium between the oxy state (oxygen-bound form, HbFe2+-O2), and deoxy state (oxygen-free form, HbFe2+) of hemoglobin, and its related functions. One cysteine residue is contained in each α globin chain (αCys104), whereas two cysteine residues can be found in each β globin chain (βCys93 and βCys112) (Figure 1). Between these three cysteine residues, αCys104 is totally hidden and βCys93 is far more reachable compared to βCys112. Therefore, βCys93 residue represents the favorite site for the binding of the tripeptide glutathione molecule in order to obtain HbSSG, even if glutathionylation at βCys112 has also been demonstrated [42][43]. This conjugation occurs in response to oxidative stress and serves as a protective mechanism against oxidative stress conditions. Attachment of glutathione to hemoglobin endows it with antioxidant properties, enabling it to scavenge harmful ROS and thwart oxidative damage to cellular components [44]. In the glutathionylation of hemoglobin (HbSH) to glutathionyl hemoglobin (HbSSG), a first possibility is its direct oxidation by ROS to the formation of the β-93-cysteine thiyl radical (Hb-S•) or the β-93-cysteine sulfenic acid (Hb-SOH) that reacts with GSH leading to HbSSG (Figure 1, mechanisms 1 and 2). Another possibility is when hemoglobin acts as a buffer scavenger of oxidized glutathione in its transient form (glutathione sulfenic acid, GS-OH), yielding the mixed disulfide protein HbSSG (Figure 1, mechanism 3) or when, under severe oxidative stress conditions, a higher ratio of GSSG to GSH is present allowing HbSH (in a deprotonated thiolate anion –S− form) to attack the sulfhydryl of GSSG (Figure 1, mechanism 4).
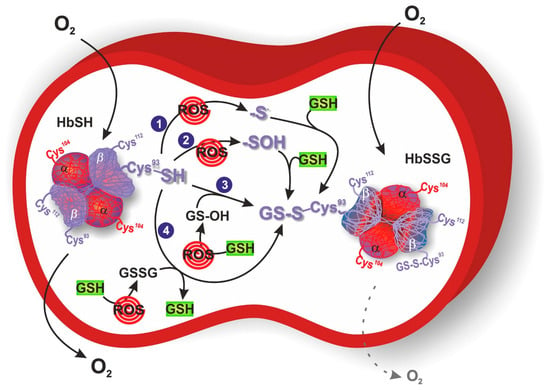
Figure 1. Response of erythrocytes to transient and chronic conditions of oxidative stress via glutathionyl hemoglobin (HbSSG) formation. In mechanism 1 and 2, oxygen reactive species (ROS) catalyze the formation of Hb β-93-cysteine thiyl radical or sulfenic acid, respectively. These intermediates react with reduced glutathione (GSH) to generate HbSSG. In mechanism 3, ROS oxidize GSH to the sulfenic acid form, which reacts with Hb β-93-cysteine to generate HbSSG. In mechanism 4, ROS increase oxidized glutathione (GSSG) concentration that can be attacked by Hb β-93-cysteine in its thiolate anion form. O2 dotted arrow represents the reduced oxygen delivery of HbSSG to targeted tissues. Hemoglobin is represented with its glutathionylation sites (αCys104, βCys93 and βCys112), where βCys93 corresponds with the favorite site for S-glutathionylation.
4. Glutathionyl Hemoglobin and Methemoglobin
HbSSG could also be produced starting from the oxidation of hemoglobin to methemoglobin within intact erythrocytes, thus establishing an indirect interconnection between HbSSG and methemoglobin [45]. In particular, methemoglobin is obtained through the reaction between the Fe(II)O2-group of oxyhemoglobin and oxidant molecules, like H2O2, nitrite, nitric oxide and hydroperoxide, which induce the oxidation of hemoglobin iron from the ferrous state (Fe2+) to the ferric one (Fe3+) [46]. In contrast to normal hemoglobin, methemoglobin is unable to transport oxygen because the transport of oxygen needs a reversible bound between oxygen and ferrous hemoglobin, thus reducing the amount of oxygen delivered to different tissues and enhancing the potential risk of tissue hypoxia [47]. Moreover, production of methemoglobin within healthy erythrocytes promotes erythrocyte disfunctions, like peroxidative changes in their membranes [45]. Under physiological conditions, methemoglobin is produced at a 3%/day rate since, although the oxygenated hemoglobin is a stable molecule, it slowly undergoes an auto-oxidization process [47]. However, several protective mechanisms, like the cytochrome b5-methemoglobin reductase pathway, maintain its levels below 1% by reducing the ferric ion (Fe3+) to the ferrous ion (Fe2+). Indeed, cytochrome b5 reductase is able to convert methemoglobin to hemoglobin through the nicotinamide adenine dinucleotide, thus eliminating 95–99% of methemoglobin [48].
Investigating the connection between methemoglobin and HbSSG, it has been reported that incubation of erythrocytes with the cell membrane permeable hydroperoxide tert-Butyl-hydroperoxide (tBHP) linearly enhances the formation of methemoglobin until 10 min of incubation (4), which in turn reacts with GSSG to form HbSSG (5). Instead, the sulfhydryl reductant dithiothreitol (DTT) avoids the production of methemoglobin by tBHP, protecting the erythrocytes from peroxidative changes (6) [45].
Hemoglobin + tBHP → methemoglobin (4)
methemoglobin + GSSG → HbSSG (5)
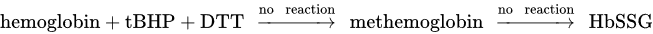
(6)
Therefore, different kinds of hemoglobin glutathionylation can occur within intact erythrocytes, including the formation of a mixed disulfide bond among GSH and normal hemoglobin, but also between cysteine residues of methemoglobin and GSSG [49].
5. Methods for the Detection of Glutathionyl Hemoglobin
In order to better highlight both the physiological and pathological role of glutathionylated proteins, including hemoglobin, several studies have been focused on the methodological systems used to quantify them.
Usually, quantification of S-glutathionylated proteins is achieved by measuring the amount of GSH released after reduction of the disulfide bond. This analysis can be carried out by spectrophotometric assays, that generally consists in the quantification of GSH after reduction of the protein mixed disulfide [50], but most of the procedures applied for the detection of S-glutathionylated proteins rely on high performance liquid chromatography (HPLC) separation to increase the sensitivity of the assay and to decrease the detection limit. The chromatographic run is generally coupled to spectrophotometric or fluorometric detection of GSH, which has been tagged with the revealing of molecules [51][52][53]. The presence of S-glutathionylated proteins can also be revealed by conventional mass spectrometry (MS) procedures. In this case, the mass difference due to GSH binding can be verified by direct electrospray ionization (ESI) measurements in intact proteins [54].
Analysis of glutathionylated proteins is challenging to perform because their concentrations could be overestimated due to GSH artefactual oxidation in the pre-analytical step, thus leading to an increase in both GSSG and glutathionylated protein levels. With the aim of preventing this experimental trouble, it has been recently developed a specific HPLC-based method by [55]. Briefly, after collection, samples have been pre-treated with an alkylating agent, then protein deglutathionylation has been induced by DTT, and the released GSH, previously marked with a fluorescent probe, has been successfully quantified by HPLC. Thiol groups of proteins are thus protected from their artificial oxidation. The main disadvantage of this method is the inability to discriminate between the various glutathionylated proteins, thus not allowing identification of each specific protein subjected to S-glutathionylation, whereas the main advantage is the possibility of accurately estimating the total quantity of glutathionylated proteins in the analyzed sample. The protocol applied by [55] can be considered for all biological samples. However, as far as blood is concerned, it appears to be particularly subject to the autoxidation of GSH, due to the presence of oxygen and iron within the heme group. Therefore, by applying this protocol in the blood, the data loses some significance, unlike the data obtained from other biological samples. Hence, investigation and development of specific techniques have become necessary for a more accurate detection and quantification of glutathionylated proteins within the blood [55].
6. Glutathionyl Hemoglobin and Diseases
Elevated levels of HbSSG have been registered in several diseases associated with oxidative stress, including chronic renal failure, DM, HLD, FRDA and IDA, probably due to the severe oxidative stress within erythrocytes of patients affected by these conditions. Among the several disorders related to oxidative damage, uremia is a pathological condition that occurs in advanced states of chronic renal failure. Patients affected by this disease may undergo hemodialysis (HD) or continuous ambulatory peritoneal dialysis (CAPD) as therapeutic strategy [56]. Uremic patients show a higher ratio of GSSG to GSH, in which GSSG represents a necessary source for the transformation of hemoglobin into HbSSG, whereas GSH within red blood cells prevents hemoglobin from being oxidized into methemoglobin. Several studies have taken into consideration patients who have undergone HD, following which there is an increase in HbSSG compared to normal subjects, probably due to the greater oxidative stress. Indeed, an excess of oxidative stress occurs after HD treatment because of both the poor or defective antioxidant defense and the activation of polymorphonuclear neutrophils through a contact system between blood and dialysis membranes, thus leading to a low concentration of GSH, an increase in GSSG and a lower activity of glutathione-dependent enzymes (GST, GR, etc.) in the red blood cells [57][58]. Other than HD, uremic patients could be treated with CAPD. Therefore, HbSSG levels have also been analyzed in this pathological condition after CAPD treatment by HPLC-ESI-MS. Concentrations of HbSSG significantly increase in CAPD patients, as well as HD patients, compared with healthy subjects. However, no statistically significant differences have been identified in HbSSG levels between HD and CAPD-treated patients [57]. To conclude, the redox balance of intracellular GSH is an important factor as it determines the good functioning of proteins with respect to oxidative stress.
7. Conclusions
Hemoglobin represents one of the most susceptible proteins to undergo the reversible PTM of S-glutathionylation following oxidative stress conditions. Levels of HbSSG show a statistically significant increase in several pathological conditions related to oxidative stress, confirming its emerging role as a clinical biomarker of severe oxidative stress but also as a pivotal pathogenic factor in different diseases. Indeed, in contrast to GSSG levels, which also increase during oxidative perturbations, HbSSG is less susceptible to enzymatic reduction, and therefore more suitable to be used as oxidative stress indicator. Moreover, S-glutathionylation of hemoglobin induces conformational modifications in its structure, resulting in a regulation of hemoglobin functions, such as an increased oxygen affinity and reduced cooperativity compared to non-modified hemoglobin, thus suggesting a role in the pathophysiology of oxidative stress-associated disorders. However, additional studies are recommended to better highlight the role of HbSSG in correlation with oxidative damage and to develop increasingly accurate, as well as reliable, techniques for its quantification in clinical investigations.
References
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28.
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550.
- Phull, A.R.; Nasir, B.; Haq, I.U.; Kim, S.J. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem. Biol. Interact. 2018, 281, 121–136.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127.
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Engin, A. Non-Alcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2017, 960, 443–467.
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679.
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal 2016, 25, 119–146.
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2017, 2017, 2818565.
- Valenzuela, P.L.; Carrera-Bastos, P.; Galvez, B.G.; Ruiz-Hurtado, G.; Ordovas, J.M.; Ruilope, L.M.; Lucia, A. Lifestyle interventions for the prevention and treatment of hypertension. Nat. Rev. Cardiol. 2021, 18, 251–275.
- Wronka, M.; Krzeminska, J.; Mlynarska, E.; Rysz, J.; Franczyk, B. The Influence of Lifestyle and Treatment on Oxidative Stress and Inflammation in Diabetes. Int. J. Mol. Sci. 2022, 23, 15743.
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Krankel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406.
- Venetsanopoulou, A.I.; Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Epidemiology of rheumatoid arthritis: Genetic and environmental influences. Expert. Rev. Clin. Immunol. 2022, 18, 923–931.
- Niwa, T. Protein glutathionylation and oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 855, 59–65.
- Smith, F.M.; Kosman, D.J. Molecular Defects in Friedreich’s Ataxia: Convergence of Oxidative Stress and Cytoskeletal Abnormalities. Front. Mol. Biosci. 2020, 7, 569293.
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514.
- Halloran, M.; Parakh, S.; Atkin, J.D. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 2013, 797914.
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox Signal 2012, 16, 543–566.
- Kuypers, F.A. Red cell membrane damage. J. Heart Valve Dis. 1998, 7, 387–395.
- Mailloux, R.J.; Gill, R.; Young, A. Chapter 13—Protein S-glutathionylation and the regulation of cellular functions. In Oxidative Stress; Academic Press: Cambridge, MA, USA, 2020; pp. 217–247.
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal 2005, 7, 348–366.
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96.
- Musaogullari, A.; Chai, Y.C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113.
- Giustarini, D.; Milzani, A.; Aldini, G.; Carini, M.; Rossi, R.; Dalle-Donne, I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid. Redox Signal 2005, 7, 930–939.
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell Mol. Med. 2004, 8, 201–212.
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal 2008, 10, 1941–1988.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal 2020, 32, 677–700.
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785.
- Mannervik, B.; Axelsson, K. Role of cytoplasmic thioltransferase in cellular regulation by thiol-disulphide interchange. Biochem. J. 1980, 190, 125–130.
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11.
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal 2013, 18, 1623–1641.
- Haendeler, J. Thioredoxin-1 and posttranslational modifications. Antioxid. Redox Signal 2006, 8, 1723–1728.
- Starke, D.W.; Chock, P.B.; Mieyal, J.J. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J. Biol. Chem. 2003, 278, 14607–14613.
- Jessop, C.E.; Chakravarthi, S.; Watkins, R.H.; Bulleid, N.J. Oxidative protein folding in the mammalian endoplasmic reticulum. Biochem. Soc. Trans. 2004, 32, 655–658.
- Mailloux, R.J.; Gill, R.; Young, A. Chapter 13—Protein S-glutathionylation and the regulation of cellular functions. In Oxidative Stress; Academic Press: Cambridge, MA, USA, 2020; pp. 217–247.
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68.
- Mitra, A.; Muralidharan, M.; Srivastava, D.; Das, R.; Bhat, V.; Mandal, A.K. Assessment of Cysteine Reactivity of Human Hemoglobin at Its Residue Level: A Mass Spectrometry-Based Approach. Hemoglobin 2017, 41, 300–305.
- Mitra, G.; Muralidharan, M.; Pinto, J.; Srinivasan, K.; Mandal, A.K. Structural perturbation of human hemoglobin on glutathionylation probed by hydrogen-deuterium exchange and MALDI mass spectrometry. Bioconjug Chem. 2011, 22, 785–793.
- Rubino, F.M. The Redox Potential of the beta-(93)-Cysteine Thiol Group in Human Hemoglobin Estimated from In Vitro Oxidant Challenge Experiments. Molecules 2021, 26, 2528.
- Murakami, K.; Mawatari, S. Oxidation of hemoglobin to methemoglobin in intact erythrocyte by a hydroperoxide induces formation of glutathionyl hemoglobin and binding of alpha-hemoglobin to membrane. Arch. Biochem. Biophys. 2003, 417, 244–250.
- Skold, A.; Cosco, D.L.; Klein, R. Methemoglobinemia: Pathogenesis, diagnosis, and management. South. Med. J. 2011, 104, 757–761.
- Mansouri, A.; Lurie, A.A. Concise review: Methemoglobinemia. Am. J. Hematol. 1993, 42, 7–12.
- Wright, R.O.; Lewander, W.J.; Woolf, A.D. Methemoglobinemia: Etiology, pharmacology, and clinical management. Ann. Emerg. Med. 1999, 34, 646–656.
- Mawatari, S.; Murakami, K. Different types of glutathionylation of hemoglobin can exist in intact erythrocytes. Arch. Biochem. Biophys. 2004, 421, 108–114.
- Rossi, R.; Cardaioli, E.; Scaloni, A.; Amiconi, G.; Di Simplicio, P. Thiol groups in proteins as endogenous reductants to determine glutathione-protein mixed disulphides in biological systems. Biochim. Biophys. Acta 1995, 1243, 230–238.
- Giustarini, D.; Dalle-Donne, I.; Colombo, R.; Petralia, S.; Giampaoletti, S.; Milzani, A.; Rossi, R. Protein glutathionylation in erythrocytes. Clin. Chem. 2003, 49, 327–330.
- Meredith, M.J. Analysis of protein-glutathione mixed disulfides by high performance liquid chromatography. Anal. Biochem. 1983, 131, 504–509.
- Paroni, R.; De Vecchi, E.; Cighetti, G.; Arcelloni, C.; Fermo, I.; Grossi, A.; Bonini, P. HPLC with o-phthalaldehyde precolumn derivatization to measure total, oxidized, and protein-bound glutathione in blood, plasma, and tissue. Clin. Chem. 1995, 41, 448–454.
- Niwa, T.; Naito, C.; Mawjood, A.H.; Imai, K. Increased glutathionyl hemoglobin in diabetes mellitus and hyperlipidemia demonstrated by liquid chromatography/electrospray ionization-mass spectrometry. Clin. Chem. 2000, 46, 82–88.
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Rossi, R. Measurement of S-glutathionylated proteins by HPLC. Amino Acids 2022, 54, 675–686.
- Mandal, A.K.; Woodi, M.; Sood, V.; Krishnaswamy, P.R.; Rao, A.; Ballal, S.; Balaram, P. Quantitation and characterization of glutathionyl haemoglobin as an oxidative stress marker in chronic renal failure by mass spectrometry. Clin. Biochem. 2007, 40, 986–994.
- Takayama, F.; Tsutsui, S.; Horie, M.; Shimokata, K.; Niwa, T. Glutathionyl hemoglobin in uremic patients undergoing hemodialysis and continuous ambulatory peritoneal dialysis. Kidney Int. Suppl. 2001, 78, S155–S158.
- Naito, C.; Kajita, M.; Niwa, T. Determination of glutathionyl hemoglobin in hemodialysis patients using electrospray ionization liquid chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 121–124.

