Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Swati Garg | -- | 3577 | 2024-01-18 17:20:03 | | | |
| 2 | Lindsay Dong | Meta information modification | 3577 | 2024-01-19 04:28:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Garg, S.; Ni, W.; Griffin, J.D.; Sattler, M. Chimeric Antigen Receptor T Cell Therapy in AML. Encyclopedia. Available online: https://encyclopedia.pub/entry/54063 (accessed on 09 August 2026).
Garg S, Ni W, Griffin JD, Sattler M. Chimeric Antigen Receptor T Cell Therapy in AML. Encyclopedia. Available at: https://encyclopedia.pub/entry/54063. Accessed August 09, 2026.
Garg, Swati, Wei Ni, James D. Griffin, Martin Sattler. "Chimeric Antigen Receptor T Cell Therapy in AML" Encyclopedia, https://encyclopedia.pub/entry/54063 (accessed August 09, 2026).
Garg, S., Ni, W., Griffin, J.D., & Sattler, M. (2024, January 18). Chimeric Antigen Receptor T Cell Therapy in AML. In Encyclopedia. https://encyclopedia.pub/entry/54063
Garg, Swati, et al. "Chimeric Antigen Receptor T Cell Therapy in AML." Encyclopedia. Web. 18 January, 2024.
Copy Citation
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy that is often associated with relapse and drug resistance after standard chemotherapy or targeted therapy, particularly in older patients. Hematopoietic stem cell transplants are looked upon as the ultimate salvage option with curative intent. Adoptive cell therapy using chimeric antigen receptors (CAR) has shown promise in B cell malignancies and is being investigated in AML.
acute myeloid leukemia (AML)
chimeric antigen receptor (CAR)
adoptive cell therapy (ACT)
1. Introduction
Immune cells work in concert to fight off foreign pathogens, eliminate aberrant cells, or contribute to wound healing. During malignant transformation, this harmony between immune cells can be disrupted, and cancer cells escape detection by the body’s immune system. While innate immune cells are mainly involved in warding off microbial infections and parasites, they may also target some tumor cells [1]. The adaptive immune system is involved in the recognition of processed intracellular antigens, antigen clearance, memory response, and immune regulation through B cells and T cells. The association between the immune state of a person and the progression of a malignant manifestation has been known for a long time. The concept of immune therapy in cancer was first documented in the late 19th century, when tumor regression was observed in some sarcoma patients after erysipelas infection [2]. The importance of an active immune system in cancer surveillance became prominent in the 1960s after the discovery of the major effectors of the immune system. Conversely, numerous reports were published of an increased incidence of cancer in patients with immune deficiencies [3][4]. The effect of adoptive cell transfer (ACT) on tumor burden was first observed in patients who received allogeneic hematopoietic stem cell transplant (HSCT) for the treatment of hematological malignancies and subsequently showed immune reconstitution along with reduced leukemia burden. Since then, several different types of adoptive cell transfer of immune cells have been developed as treatment strategies against different types of malignancies [5].
In acute myeloid leukemia (AML), myeloid lineage blood-forming cells are arrested at different developmental stages (often referred to as ‘blasts’) and grow uncontrollably, which results in rapid hematopoietic insufficiency and bone marrow failure. AML is a heterogeneous cancer that generally affects older adults, with a median age of diagnosis of 65 years. Due to its diverse biology, wide spectrum of chromosomal aberrations, and mutational landscape, the clinical outcomes after standard induction treatment with daunorubicin and cytarabine are somewhat dismal, especially for patients older than 65 years of age, where the 5-year overall survival for this population is still below 10–15% [6]. For a subset of AML patients, HSCT after frontline induction chemotherapy has proven to be the most effective treatment, and tumor-reactive T cells from the donor are believed to be a major co contributors to this success [7].
There has been an intense effort to develop drugs that directly target the oncogenes that cause AML, and some successes have been noted. Acute promyelocytic leukemia (APL) with a PML-RARα fusion oncogene is so far the only AML subgroup where such targeted agents can be used successfully without concomitant chemotherapy. In APL, all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO), two drugs that inhibit PML-RARα, can be used without standard chemotherapy for disease management with a high rate of complete remission and cure. Other new targeted agents have been shown to have survival benefits in selected AML subsets when added to standard chemotherapy regimens, including inhibitors of mutant FLT3 (midostaurin, sorafinib, gilteritinib, quizartinib, and others), IDH1 (ivosidenib), and IDH2 (enasidenib). These and other reagents in clinical development are often combined with the BCL2-targeting venetoclax to improve the induction of cell death in AML.
2. How Do Immune Cells Work?
The immune system comprises myeloid (monocytes, macrophages, and granulocytes), lymphoid (B, T, and natural killer cells (NK)), and dendritic cells (DC). As the first response, common molecular patterns on the surface of pathogenic bacteria or cells are recognized by the cells of the innate immune system, such as monocytes, DCs, granulocytes, NK, and innate lymphoid cells, which lead to the activation of immune components, including adaptive immune cells such as B and T cells. While monocytes, macrophages, granulocytes, and B cells are generally involved in phagocytosis of pathogens and antigen presentation, cytotoxic T cells and NK cells have direct cytolytic functions. The three major lymphoid cells have specific functions: B cells are specialized APCs that secrete antigen-neutralizing antibodies and are involved in immune activation. NK cells can detect a variety of stress-induced self and infectious non-self ligands to activate cytotoxicity through the release of lytic granules in target cells and the release of cytokines for immune activation. T cells have immune-activating, regulatory, and direct cytolytic roles in immune reactions [8][9][10][11].
T cells play a central role in the immune system by establishing successful cell-based immune responses. They recognize antigen fragments presented by the major histocompatibility complexes (MHC) on the surface of antigen-presenting cells (APCs) through the T cell receptor (TCR) complex. Most T cells in the periphery express TCR made of alpha/beta glycoproteins, and a minority of T cells express gamma/delta TCR. These glycoproteins belong to the immunoglobulin (Ig) superfamily, and like the Igs, TCRs have constant and variable regions, transmembrane and cytoplasmic domains, and the genes encoding TCRs undergo similar splicing events as those of B cell membrane immunoglobulins. However, unlike Igs, TCRs can only identify an antigen when presented with MHC. Of note, in CAR T cells, the MHC restriction is removed by substituting the TCR recognition function with an Ig-like element. The TCR complex itself consists of non-covalently linked CD3 chains that form heterodimers comprising epsilon-gamma (εγ), epsilon-delta (εδ), and zeta-zeta (ζζ) signaling chains that form an octamer TCR complex [12][13]. A mature T cell in the periphery expresses a clone-specific TCR complex along with one of the two co-receptors, CD4 or CD8, that determine its MHC restriction [14][15]. CD4 T cells recognize mainly exogenous, or pathogen-derived antigens such as bacterial peptides presented in the MHC II complex on antigen-presenting cells. T helper or CD4 T cells orchestrate immuno-protective and regulatory functions by releasing inflammatory cytokines, chemokines, trans phagocytosis, and recruitment of other immune cells [16][17]. CD8 T cells mainly recognize self-derived antigens presented with MHC class I complexes on the antigen-presenting cells [18][19]. Since MHC I is expressed on almost all nucleated cells, virally infected and tumor-associated antigens are efficiently captured via CD8 T cells, which have direct cytolytic function upon activation. There are additional T cell types defined by their functionality, differential antigens, and cytokines. Figure 1A depicts the general concept of antigen recognition by CD4 and CD8 T cells. While TCR and co-receptor engagement are sufficient for the initial priming, antigen recognition, and thymic selection, a costimulatory signal through CD28 is required for optimal T cell activation. The B7 family of immunomodulatory receptors, including CD28 ligands B-7, CD80 (B7-1) and CD86 (B7-2) are expressed on APCs and play an important role at immunological synapses (IS). CD28 mediates the full activation of T cells by organizing the membrane rafts to IS through activation of a series of signaling events that include phosphorylation of key immunoreceptor tyrosine-based activation motifs (ITAM) [20].
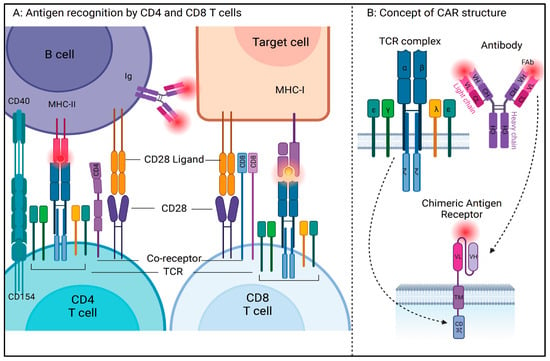
Figure 1. (A) Antigen recognition by CD4 and CD8 T cells in conjunction with MHC complex expressed on antigen-presenting cells. (B) Concept of CAR structure: To circumvent the MHC restriction of antigen recognition by T cells, synthetic receptor containing the complementarity-determining regions (CDR) on variable light and heavy (vH and vL) chains of monoclonal antibodies are cloned into a single variable fragment (scFv), which is then attached to the transmembrane and intracellular signaling domains of T cell receptor components via hinge region (created with BioRender (https://www.biorender.com/, accessed on 17 July 2023)).
3. How Do Cancer Cells Evade the Immune System?
According to the cancer immunosurveillance hypothesis and its addendums, immune cells can recognize and regulate the growth of transformed malignant cells, but they can also allow malignant cells with low immunogenicity to outgrow. Cancer cells are known for immunoediting through several mechanisms at multiple levels. As discussed earlier, for a T cell to recognize a self-transformed cell, an antigen has to be presented through the MHC-I complex; downregulation of MHC-I and other components of antigen presentation pathways serve as an important immune escape mechanism for several tumors [21]. Cancer cells can also escape immune recognition by lowering their antigenicity, whereby unless the transformed cell expresses a neo or aberrant antigen at high levels, it cannot be recognized by the cells of the immune system. Cancer cells can achieve a reduction in immunogenicity by dampening T cell activation and inducing regulatory function via overexpression of immune checkpoint ligands such as PD-L1, MHC-II, FGL1, Galectin-9, and HMGB1 to engage PD1, LAG3, and TIM3 on T cells [22][23][24]. The cytokine composition of the tumor microenvironment can play a repressive role for immune helper cells such as dendritic cells (DCs) by releasing IL10, prostaglandin-2, TGF-beta, and indoleamine 2,3-dioxygenase (IDO) [25]. Type I interferon-induced epigenetic reprogramming for increased stemness and immune escape in cancer cells is yet another mechanism with which cancer cells can render normal immune cells non-functional against them [26][27]. This is not a comprehensive list of all immune escape mechanisms, but in general, immunotherapy and CAR cell therapy attempt to circumnavigate many of these obstacles to arrive at an activated anti-tumor-specific state.
4. What Is a Chimeric Antigen Receptor (CAR) T Cell?
CAR T cells are genetically engineered T cells that express an exogenous, synthetic receptor able to recognize a specific antigen in the absence of MHC restriction, leading to optimized T cell activation for effective target cell killing. The concept of CAR T cells first came to light in 1987 when Dr. Zelig Eshhar retrovirally infected T cells to express an engineered TCR with the goal of killing tumor cells. In the late 1980s, several groups, including Dr. Steven Rosenberg at the NCI, Dr. Carl June at Penn Medicine, Dr. Michel Sadelain at MSKCC, Dr. Dario Campana at St. Jude, and others, tried to develop more effective CAR T cells. By the end of the 1990s, the groups of Dr. Sadelain and Dr. June showed the translational optimization of engineered T cells for clinical application, crediting Dr. June as the pioneer of CAR T therapy [28][29].
5. What Is the Structure of a CAR?
Conceptually, prototype CAR T receptors require four main structural components in order to sustain an effective T-cell-mediated immune response [30] (Figure 1B), including the following:
-
Antigen-Binding Domain: The discovery of monoclonal antibodies in the 1970s played an important role in the conceptualization of antigen specificity using complementary determining regions of variable and constant regions of immunoglobulins [31]. In a CAR antigen, the antigen-binding domain is a single-chain variable fragment (scFv), derived from variable light (vL) and heavy chains (vH), and a flexible linker of the antigen-specific monoclonal antibody. The usage of antibody-mediated antigen recognition systems allows the CAR T cell to circumvent MHC restriction;
-
Hinge Region: This region connects the antigen-binding domain to the transmembrane domain and affects the overall steric conformation of the CAR to the antigen. These can be of various lengths and are generally derived from the sequence of T cell coreceptors such as CD8, CD28, or immunoglobulins. Shorter extracellular domains increase the potential for CAR T activation, whereas lengthening the CAR antigen diminishes CAR T activation [32];
-
Transmembrane Domain: This domain is the region that anchors the CAR to the T cell membrane. Generally, TM domains are derived from amino acid sequences of T cell coreceptors such as CD4, CD8, CD3zeta, and CD28 and are reported to be involved in cytokine release and cell death, apart from their overall stability. The stability of CAR and its expression on the T cell membrane is affected by the transmembrane domain, whereas the hinge domain is critical in the regulation of signaling threshold [33];
-
Intracellular Signaling Domain: This domain, also called the costimulatory (CM) domain, transduces the signaling cascade and is involved in T cell activation after successful antigen recognition. This is the domain that contains the necessary ITAMs for downstream signaling cascade activation. Because of its role in cell stimulation, cytokine release, and activation-induced cell death, the intracellular domain has been the most focused of all the regions over the years through multiple generations of CAR antigens.
6. How Did the CAR Design Evolve?
Over the years, CAR designs have emerged to improve efficacy, reduce toxicity, increase safety, and overcome the limitations posed by the tumor microenvironment, tumor immune evasion, and relapse. Various CAR designs have shown their promise in experimental and clinical settings. The basic structural concept of the CAR molecule remains the same, but different domains and sequences have shown differential activity and efficacy. The first-generation CAR incorporated a single intracellular domain derived from the zeta chain of CD3 and lacked persistent in vivo proliferation. To overcome the challenge of limited in vivo proliferation, in the second-generation CAR, one additional costimulatory domain was added to the intracellular region, which was derived from either CD28 or 4-1BB. These CARs showed better proliferation in the absence of exogenous signals. To achieve optimal activation, additional ITAMs were designed in the third-generation CAR, where two additional intracellular domains from the receptors, such as CD28, 4-1BB, CD27, CD40, OXO-4, DAP-12, or ICOS, were used [34][35].
7. What Are the Challenges and Strategies to Overcome Shortcomings in CAR Design?
Despite the seemingly straight-forward immune reaction required to kill tumor cells using engineered CAR T cells, challenges arise whereby CAR T cells cannot recognize escaped tumor cells that relapse in either the same or altered phenotype, or CAR treatment results in acute toxicity leading to life-threatening conditions. Therefore, numerous strategies are utilized to optimize CAR design to address specific challenges (Figure 2).
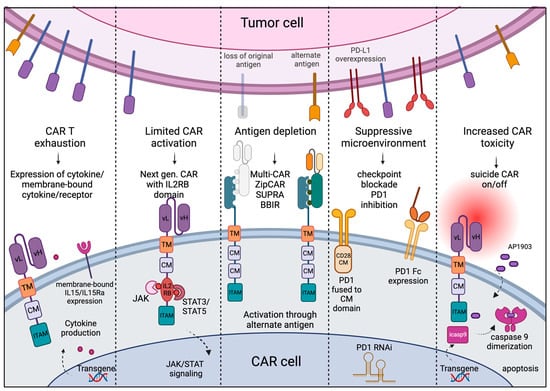
Figure 2. Challenges and approaches: Cell exhaustion, limited activation, and increased off-target toxicity of CAR cells can be regulated by providing cytokine transgenes in armored CARs, adding IL2-receptor-binding domain to enhance JAK/STAT signaling for T cell activation, and off-signaling in response to dimerizing agents to dampen the toxicity. Use of universal CAR design can circumvent the problem of antigen depletion and outgrowth of tumor population with alternate antigens, and use of immune checkpoint blockade can overcome the suppressive tumor microenvironment. (Created with BioRender.)
8. CAR Clinical Trials in AML
An efficient and specific ‘on-target’ CAR-mediated tumor cell killing depends on the antigen against which the CAR is generated, the ability of the CAR cell to reach and recognize the malignant cells, and the microenvironment of the given tumor. Since AML is a bone marrow and blood disorder, low circulation of the CAR T cells is not likely to be a challenge, and CAR T efficacy is in large part limited to the features of the antigen. It is important that the antigen against which the CAR is generated (a) expresses abundantly and differentially on the surface of tumor cells compared to the normal cells of the same tissue, (b) shows minimal to no expression on the surface of normal cells across different tissues, and (c) does not express on the surface of T cells. So far, CAR T therapy has been most successful in the treatment of B cell malignancies, and the first recipient of CD19-targeting CAR T therapy was recently reported to be in complete remission (CR) for a decade [36]. The six US FDA-approved CAR products are for B cell malignancies, where four of those products target CD19 and two are against B Cell Maturation Antigen (BCMA) [37]. Both CD19 and BCMA are B cell differential antigens. B cells are specialized APCs that produce neutralizing antibodies. In the event of B cell depletion, immunoglobulins can be externally given, and the myeloid cells can work as bona fide APCs without causing generalized hemotoxicity. The availability of commercial CAR products, large amounts of real-world data, and clinical guidelines prove the efficacy of CAR T therapy in relapsed/refractory (R/R) B-ALL [38]. As of June 2023, 92 clinical trials are listed on the clinicaltrials.gov website that use CAR treatment in AML. These trials include about 24 antigens, which can be divided into groups based on their expression patterns (overexpressed myeloid, repurposed or aberrant lymphoid, or neoantigens) and have been extensively reviewed in the literature [7][34][39][40][41][42]. The expression of these antigens at the transcript level in different tissues (Figure 3A; data from the human proteome atlas [43] and GTEx consortium [44], downloaded from proteinatlas.org and prepared using iDEP [45]) shows potential antigen distribution in normal cells. The expression of these antigens at mRNA levels in sorted lineage fractions from 12 healthy bone marrow and 9 AML samples [46] (Figure 3B) suggests only limited specificity for leukemic cells for WT1, but not for other antigens.
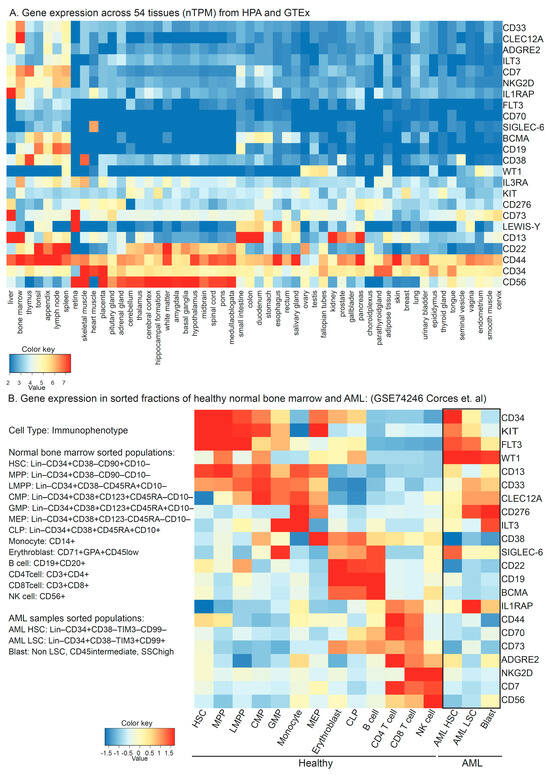
Figure 3. (A) Transcript-level expression of CAR targets in AML across 54 normal tissues in a consensus dataset retrieved at https://www.proteinatlas.org/ (accessed on 1 June 2023). (B) Expression of potential CAR targets at the mRNA level in sorted cell populations of normal healthy marrows from 12 donors and AML samples from 9 patients in single-cell RNA sequencing experiment submitted at gene expression omnibus GSE74246. HSC: Hematopoietic Stem Cells, MPP: Multipotent Progenitor, LMPP: Lymphoid–Myeloid Primed Progenitors, CMP: Common Myeloid Progenitors, GMP: Granulocyte–Monocyte Progenitors, MEP: Megakaryocyte–Erythrocyte Progenitors, and CLP: Common Lymphoid Progenitor.
9. Improving CAR T Cell Therapy One Step at a Time
At first glance, a common theme of CAR therapy in AML is characterized by the variety of target antigens against which the receptor is developed. This may reflect the heterogeneity within the disease or a lack of prominent AML-specific antigens that can serve as targets. It appears that the field is still trying to provide an answer to this problem in multiple small trials rather than large-scale, multi-centric clinical trials. However, lessons can be learned from these studies. A meta-analysis that included 57 patients from 13 original reports (10 trials and 3 case reports) of CAR therapy for intervention in R/R AML reported complete remission (CR) in 38.5% (22 of 57, estimated pooled incidence 49.5%) patients. However, only one patient was reported to be free of cytogenetic disease for more than 20 months, and most of the patients either relapsed within six months or were subjected to further HCT if they remained negative for minimal residual disease (MRD) for up to four weeks after CAR infusion. Due to the huge variation in the endpoint set up in different studies and the small number of patients per trial, a cumulative overall survival could not be calculated. Pooled toxicity incidence surpassed the fraction of complete response as CRS was reported in 43.8% (25 of 57, pooled incidence of 54.4%) patients, and two deaths were reported due to grade IV Graft-versus-Host Disease (GvHD). The meta-analysis also included findings from an activated NK-92 clinical trial without CAR target NCT00900809, where disease progression was reported in all the patients [47]. This analysis revealed five major challenges across the included studies—(a) disease heterogeneity: different studies have different inclusion criteria, patients with different mutational burdens, and previous treatments; (b) lack of a universal target: although CD33, CLL1, CD123, and NKG2D were the main targets, most studies reported less than 10 patients treated with CAR targeted against one antigen, which makes it difficult to draw conclusions about the efficacy of a universal CAR product; (c) toxicity: due to the lack of a ‘safe target’, almost half of the patients developed CRS, five reported the development of immune effector cell-associated neurotoxicity syndrome (ICANS), and other toxicities were reported in individual studies; (d) limited off-the-shelf option: although allogeneic T cell products seem like a lucrative option, the meta-analysis observed that patients who developed severe GvHD were given allogeneic CAR T products; and (e) modest response due to CAR exhaustion: resulting in CAR therapy only being used as a bridge to HSCT rather than a curative intervention.
Some additional lessons can also be drawn from recently completed trials. In March 2023, Celyad Oncology (Belgium) released results from their multi-center THINK study, with the infusion of an autologous CAR T product (CYAD-01) against NKG2D given to 16 R/R AML, MDS, and MM patients who had received at least one line of previous treatment [48]. Ligands for NKG2D are expressed on a variety of malignant cells and are generally absent from normal cells. As expected, the study reported no myelosuppression, no neurotoxicity, and limited cytokine release syndrome (CRS). However, two major challenges of this trial were (a) manufacturing failure in patients with high leukemic burden and (b) limited response due to the lack of CAR persistence. Prior lymphodepletion and bridging therapy to overcome the cytokine sink caused by the high number of blasts and optimization of the CAR protocol to select functional T cells instead of memory cells have been suggested to overcome these challenges [49].
10. Conclusions
Although there are several challenges that remain to be addressed before CAR therapy can be used as a singular intervention in AML, the potential of CAR treatment, especially for R/R AML cases with poor prognostic mutations, complex karyotypes, and higher disease burden, is undeniable. Alternatively, CAR therapy could also be established as bridge therapy before HSCT. Due to the paucity of AML-exclusive targets, myeloablation remains a major concern for CAR therapy in AML. This is true in particular for CAR targeting CD33 and CD123, both of which are broadly expressed in the myeloid compartment. Strategies to control CAR therapy through inducible receptor activity would be helpful here and are likely to be required in AML. Another interesting strategy are universal CAR systems that would allow for the use of multiple receptors to adapt to the patients’ unique antigen profiles. Receptors could be used in combination to delay or avoid resistance. As pointed out above, there are also some differences between T cells and NK cells, but the jury is still out on whether either system is truly more advantageous to carry the CAR treatment.
References
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. eLife 2014, 3, e04177.
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965.
- Mortaz, E.; Tabarsi, P.; Mansouri, D.; Khosravi, A.; Garssen, J.; Velayati, A.; Adcock, I.M. Cancers Related to Immunodeficiencies: Update and Perspectives. Front. Immunol. 2016, 7, 365.
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250.
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J.P. Adoptive T Cell Immunotherapy for Cancer. Rambam Maimonides Med. J. 2015, 6, e0004.
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41.
- Hansrivijit, P.; Gale, R.P.; Barrett, J.; Ciurea, S.O. Cellular therapy for acute myeloid Leukemia–Current status and future prospects. Blood Rev. 2019, 37, 100578.
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510.
- Ochando, J.; Mulder, W.J.M.; Madsen, J.C.; Netea, M.G.; Duivenvoorden, R. Trained immunity—basic concepts and contributions to immunopathology. Nat. Rev. Nephrol. 2022, 19, 23–37.
- Ruf, B.; Greten, T.F.; Korangy, F. Innate lymphoid cells and innate-like T cells in cancer—at the crossroads of innate and adaptive immunity. Nat. Rev. Cancer 2023, 23, 351–371.
- De Maria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56, Erratum in 2019, 576, E3.
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619.
- Mariuzza, R.A.; Agnihotri, P.; Orban, J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J. Biol. Chem. 2019, 295, 914–925.
- Van Laethem, F.; Tikhonova, A.N.; Singer, A. MHC restriction is imposed on a diverse T cell receptor repertoire by CD4 and CD8 co-receptors during thymic selection. Trends Immunol. 2012, 33, 437–441.
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478.
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Osuna-Pérez, J.; Torres-Torresano, M.; Zorita, V.; Martínez-Riaño, A.; Boccasavia, V.; Borroto, A.; del Hoyo, G.M.; González-Granado, J.M.; et al. Conventional CD4+ T cells present bacterial antigens to induce cytotoxic and memory CD8+ T cell responses. Nat. Commun. 2017, 8, 1591.
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216.
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836.
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528.
- Zumerle, S.; Molon, B.; Viola, A. Membrane Rafts in T Cell Activation: A Spotlight on CD28 Costimulation. Front. Immunol. 2017, 8, 1467.
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568.
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692.
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185.
- Shi, A.-P.; Tang, X.-Y.; Xiong, Y.-L.; Zheng, K.-F.; Liu, Y.-J.; Shi, X.-G.; Lv, Y.; Jiang, T.; Ma, N.; Zhao, J.-B. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2022, 12, 785091.
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695.
- Musella, M.; Guarracino, A.; Manduca, N.; Galassi, C.; Ruggiero, E.; Potenza, A.; Maccafeo, E.; Manic, G.; Mattiello, L.; Rehim, S.S.A.; et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat. Immunol. 2022, 23, 1379–1392.
- Costoya, J.A.; Arce, V.M. Cancer cells escape the immune system by increasing stemness through epigenetic reprogramming. Cell. Mol. Immunol. 2022, 20, 6–7.
- Styczyński, J. A brief history of CAR-T cells: From laboratory to the bedside. Acta Haematol. Pol. 2020, 51, 2–5.
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73.
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69.
- Alkan, S.S. Monoclonal antibodies: The story of a discovery that revolutionized science and medicine. Nat. Rev. Immunol. 2004, 4, 153–156.
- Xiao, Q.; Zhang, X.; Tu, L.; Cao, J.; Hinrichs, C.S.; Su, X. Size-dependent activation of CAR-T cells. Sci. Immunol. 2022, 7, eabl3995.
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182.
- Hofmann, S.; Schubert, M.-L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 8, 200.
- Akhoundi, M.; Mohammadi, M.; Sahraei, S.S.; Sheykhhasan, M.; Fayazi, N. CAR T cell therapy as a promising approach in cancer immunotherapy: Challenges and opportunities. Cell. Oncol. 2021, 44, 495–523.
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371.
- Chen, Y.-J.; Abila, B.; Kamel, Y.M. CAR-T: What Is Next? Cancers 2023, 15, 663.
- Myers, R.M.; Shah, N.N.; Pulsipher, M.A. How I use risk factors for success or failure of CD19 CAR T cells to guide management of children and AYA with B-cell ALL. Blood 2023, 141, 1251–1264.
- Schorr, C.; Perna, F. Targets for chimeric antigen receptor T-cell therapy of acute myeloid leukemia. Front. Immunol. 2022, 13, 1085978.
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res. Ther. 2021, 12, 465.
- Koedam, J.; Wermke, M.; Ehninger, A.; Cartellieri, M.; Ehninger, G. Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr. Opin. Hematol. 2022, 29, 74–83.
- Vishwasrao, P.; Li, G.; Boucher, J.C.; Smith, D.L.; Hui, S.K. Emerging CAR T Cell Strategies for the Treatment of AML. Cancers 2022, 14, 1241.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419.
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610.
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 1–24.
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203.
- Shahzad, M.; Nguyen, A.; Hussain, A.; Ammad-Ud-Din, M.; Faisal, M.S.; Tariq, E.; Ali, F.; Butt, A.; Anwar, I.; Chaudhary, S.G.; et al. Outcomes with chimeric antigen receptor t-cell therapy in relapsed or refractory acute myeloid leukemia: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1152457.
- Sallman, D.A.; Kerre, T.; Havelange, V.; Poiré, X.; Lewalle, P.; Wang, E.S.; Brayer, J.B.; Davila, M.L.; Moors, I.; Machiels, J.-P.; et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): Haematological cohorts of the dose escalation segment of a phase 1 trial. Lancet Haematol. 2023, 10, e191–e202.
- Chua, C.C.; Cheok, K.P.L. Taking a step forward in CAR T-cell therapy for acute myeloid leukaemia and myelodysplastic syndrome. Lancet Haematol. 2023, 10, e161–e162.
More
Information
Subjects:
Immunology; Hematology; Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
667
Revisions:
2 times
(View History)
Update Date:
19 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No