From an evolutive perspective, tumor cells endure successive turnover upon stress conditions and pressure to adapt to new environments. These cells use exceptional communication skills to share biological information to “survive upon every metabolic cost”. The tumor microenvironment (TME) is a miscellaneous collection of cells, factors, and extracellular vesicles (EVs). EVs are small lipid bilayer-delimited particles derived from cells with sizes ranging from 100 to 1000 nm. Exosomes (<160 nm) are the minor subtype of EVs, originating from the endosomal pathways. The TME also contains “giant” vesicles, microvesicles (100–1000 nm, MV), originated from membrane blebbing. EVs can act as intercellular communication mediators, contributing to many biological processes, by carrying different biomolecules, such as proteins, lipids, nucleic acids, and metabolites. EV secretion can promote either tumor cell survival or manage their stress to death. Tumor-derived EVs transfer adaptative stress signaling to recipient cells, reprograming these cells. Heat shock proteins (HSP) are prominent stress response regulators, specifically carried by exosomes. HSP-loaded EVs reprogram tumor and TME cells to acquire mechanisms contributing to tumor progression and therapy resistance. The intercellular communication mediated by HSP-loaded EVs favors the escape of tumor cells from the endoplasmic reticulum stress, hypoxia, apoptosis, and anticancer therapies. Extracellular HSPs activate and deactivate the immune response, induce cell differentiation, change vascular homeostasis, and help to augment the pre-metastatic niche formation.
1. Extracellular Vesicle (EV)-Mediated Stress Propagation during Tumor Development and Progression
Cells release EVs because of their physiology and pathophysiology. EVs can be classified into three main subtypes
[1] according to their biogenesis and size. Apoptotic bodies are generated by membrane disintegration after injuries and cell death activation, producing vesicles having a diameter ranging from 1 to 5 µM. Microvesicles are particles generated by the direct outward budding of the plasma membrane of viable cells, having vesicles in a size range of around 50 nm to 1000 nm in diameter. Finally, exosomes are generated by the endosomal sorting complex required for the transport (ESCRT) system composed of different proteins able to interact with other proteins and promote the formation of intraluminal vesicles. Exosome formation starts after the invagination of the plasma membrane and the formation of an early-sorting endosome (ESE). The ESE contains cell-surface and soluble proteins associated with the extracellular milieu in addition to content from the trans-Golgi network and endoplasmic reticulum. Late-sorting endosomes (LSEs) are matured ESEs that generate multivesicular bodies (MVBs) containing intraluminal vesicles. MVBs form by double invagination of the plasma membrane and can later fuse with lysosomes or autophagosomes to be degraded. Release of exosomes occurs after the fusion of MVBs to the plasma membrane; these exosomes have a size of around 40 to 160 nm in diameter
[2].
Studies have been conducted to investigate the role of EVs in the transfer of the stress tolerance phenotype in different cancer models. Hypoxia leads to EV release and/or a higher cargo loading per vesicle, and transfers a hypoxic tolerance phenotype to TME, as well as promoting pro-tumoral effects, including induced proliferation, migration, angiogenesis, and immunomodulation
[3]. The main cellular hypoxia effect is protein-folding instability with damaged and misfolded protein accumulation. Proteostasis instability affects the endoplasmic reticulum (ER), triggering a specific cellular state known as ER stress and the unfolded protein response (UPR) to restore homeostasis. Mahadevan et al. demonstrated that ER stress can be transmitted from cancer cells to bone marrow-derived myeloid cells, a phenomenon known as transmissible ER stress
[4]. This communication was confirmed to be organized by EVs, which was firstly attributed to cancer cell soluble factors
[5], and compelling evidence from cancer cells submitted to ER stress inducers demonstrates that this insult increases EV secretion
[6][7].
EV-mediated remote stress preconditioning has identified several EV cargos, including mRNA, microRNA, and proteins that impact HIF-1α-, UPR-, angiogenesis-, and autophagy signaling in recipient cells (reviewed in
[3]). Given the critical role of HSPs in driving the stress response, their activity is one of the main cellular pro-survival mechanisms, and it would be logical to expect the presence of these proteins in EVs. Five major mammalian HSP families have been classified according to the guideline proposed by Kampinga et al.: HSP70 superfamily (HSP70 (HSPA) and HSP110 (HSPH)), DNAJ (HSP40), small heat shock proteins (HSPB), HSPC (HSP90), and chaperonins (HSPD/E)
[8]. EVs containing diverse HSP members are passively or actively released by damaged, stressed, or dead cells. The expression of HSP90 (HSPC family) in exosomes derived from diverse normal cells was previously reported
[9][10][11][12]. Later, B cell-derived exosomes were reported to have increased levels of chaperones under heat stress conditions
[13]. Oral squamous cell carcinoma secretes HSP90-enriched EVs and promotes expression of HSP90, TRAP1, and HSP105 (HSP70 superfamily), which were correlated with poor prognosis in head and neck carcinoma patients
[14]. A mitochondrial chaperonin, HSP60 (chaperonin family), is secreted into exosomes as a regular process independent of cell death induction
[15]. A mitochondrial chaperone, GRP75/mt-HSP70 (HSP70 superfamily), is involved in EV secretion by breast cancer cells and its blockage decreases tumoral EV secretion
[16]. The release of EV-HSP70 (HSP70 superfamily) can also be enhanced immediately in plasma after cardio-exercising, which follows the return to the baseline quantitate amount of HSPs in EVs extracted from patients’ serum
[17].
2. Cancer Cell Intrinsic Mechanism Modulated by HSP-EVs That Impact Therapy Response
In addition to the transfer of HSP cargo from EVs to cancer cells, HSPs present on the external surface of EVs can interact with surface receptors of target cancer cells and contribute to resistance phenotype propagation. McCready et al. described that invasive cancer cells secrete HSP90α-EVs and also identified the pro-migratory protein plasminogen as a potential client protein of these extracellular chaperones
[18]. Tsen et al. revealed another mechanism by which HSP90α, the inducible cytosolic isoform of HSP90, can modulate cancer cell migration. In this research, they provided evidence that extracellular HSP90α binds to the subdomain II of the extracellular part of low-density lipoprotein receptor-related protein 1 (LRP-1), which signals to Akt kinases, Akt1 and Akt2, to promote cell motility
[19]. Similarly, Ono et al. showed that HSP90-EVs derived from metastatic oral cancer cells initiate epithelial-mesenchymal transition (EMT) in normal epithelial cells and promote migration and invasion of tumor cells. Moreover, these EV-driven migratory events were reversed by HSP90 depletion
[20]. Tang et al. also demonstrated that breast-cancer-derived exosomes present HSP90α on their external surface and stimulate the migration of both normal stromal cells and tumor cells in a paracrine and autocrine mechanisms
[21]. The intercellular transfer of chemoresistance mediated by HSPs-EVs was shown by Wang et al.
[22]. In their study, they demonstrated that the transfer of DNAJB8 (HSP40 family) by EVs derived from oxaliplatin-resistant cells could transfer the resistance phenotype to recipient colon cancer cells. HSP-EVs can also mediate the communication of cancer cells with other stromal cells, such as endothelial cells, and promote angiogenesis. Yukawa et al. investigated the influence of exosomes secreted from hepatocellular carcinoma cells on angiogenesis and found that HepG2-derived exosomes expressing HSP70 are incorporated by HUVEC cells and induce lumen formation
[23]. Notably, several reports have suggested a key role of HSP90 in regulating tumor angiogenesis, as multiple arms of angiogenic signaling have been described as clients of this chaperone
[24]. Feng et al. reported that HSP90 is directly associated with a smaller (165 amino acid) VEGF isoform found in the outer surface of microvesicles (MVs) isolated from MDAMB231 and SKBR3 breast cancer cells. Interestingly, this association results in a sustained activation of VEGFRs and a consequent resistance to Bevacizumab. However, HSP90 inhibitors disrupt this client–protein interaction and the release of VEGF90K from the MVs restores Bevacizumab sensitivity
[25]. HSP-EVs can also promote the activation of fibroblasts in the pre-metastatic niche (PNM). Sun et al. demonstrated that HSP60 present on the surface of EVs derived from tumor cells was crucial for EV-induced lung PMN formation. HSP60-EVs in circulation could mediate immunomodulatory effects and immune response. Colon carcinoma patients present HSP60 expression in macrophages and NK cells; at the same time, HSP-EVs are present in the blood of the patients. High levels of EVs in circulation are dependent on tumor presence because, after tumor removal, HSP60-EVSs in circulation decrease
[26]. The same scenario is found in other tumor types; in thyroid papillary carcinomas, HSP27-, HSP60-, and HSP90-EV levels in plasma decreased in number after surgical resection of the tumor
[27]. These data support the idea that HSP60-EVs in circulation may be useful to follow up in patients’ recurrence after surgical treatments, for instance.
Taken together, all these reports clearly demonstrate that HSP-EVs can contribute to tumor heterogeneity response to anticancer therapies by inducing EMT, migration, and angiogenesis. It is interesting to stress that HSP-EVs can also propagate cancer drug resistance by interacting with and modulating crucial components of the immune response.
3. Immunological Roles of Cancer HSP-EVs That Impact Therapy Response
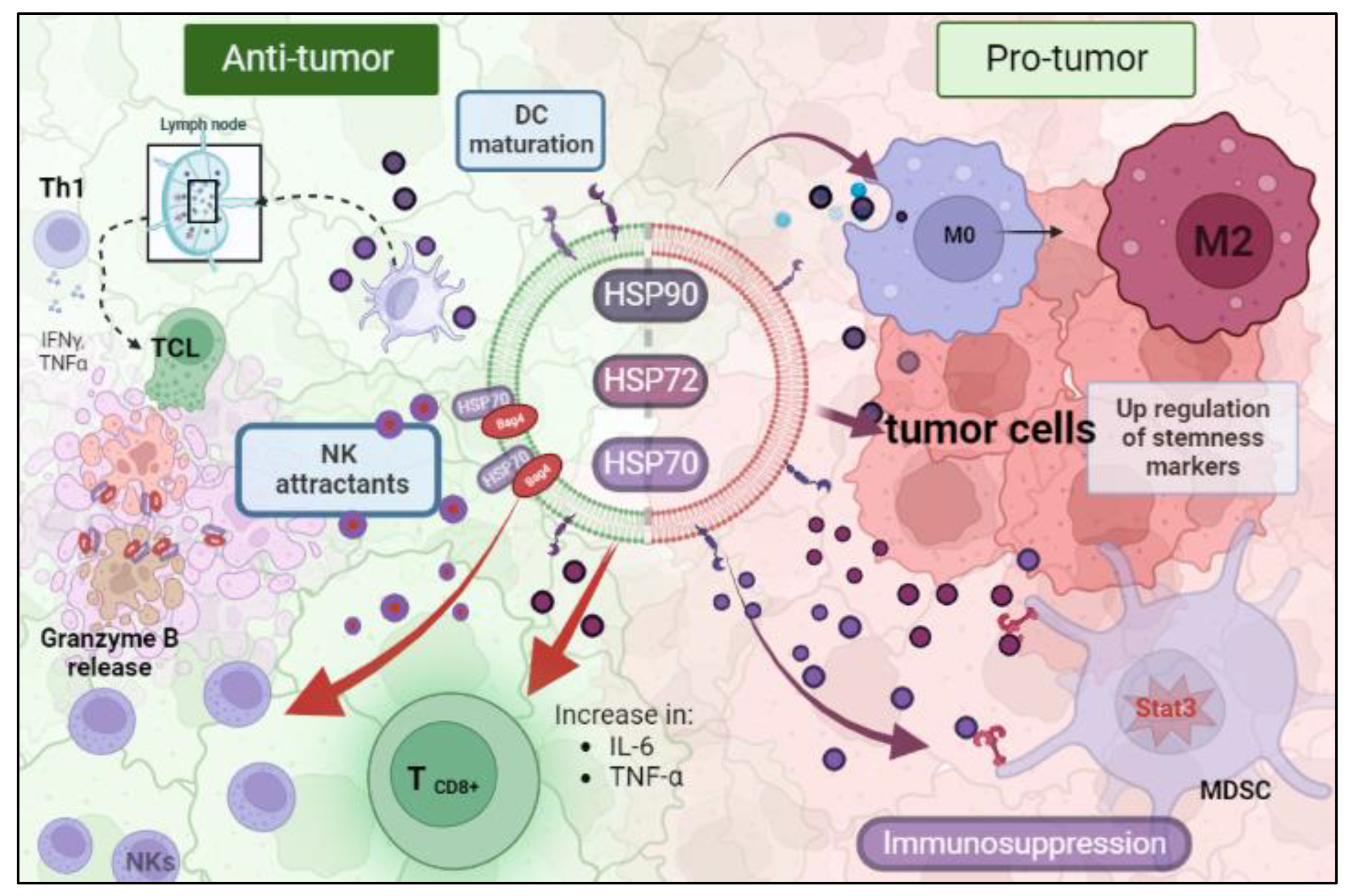
Many studies have been conducted to reveal the immunological consequences of tumoral HSP-EVs. All processes related to the immune system are tissue-context dependent on which the response is occurring. In the TME and in the presence of HSP-EVs, this idea is no different. Therefore, HSP-EVs can have an anti-tumor or pro-tumor role (
Figure 1). Chalmin et al. and Diao et al. provided a mechanistic insight linking HSP72 and HSP70 present in tumor-derived exosomes (TDEs) and tumor-induced immunosuppression, respectively. They showed that both HSP70 and HSP72 are present in TDEs and they can bind to toll-like receptor 2 (TLR2) in myeloid-derived suppressor cells (MDSCs), triggering Stat3 activation and, promoting the suppressor activity of MDSCs
[28][29]. Chalmin et al. also observed that dimethyl amiloride reduced exosome secretion and Stat3 phosphorylation in MDSCs, resulting in an enhanced cytotoxic effect on T cells under cyclophosphamide treatment
[29]. In line with this, Gobbo et al. evaluated the blockage of HSP70 and TLR2 association by using the peptide aptamer A8, which targets the extracellular domain of membrane-bound HSP70 on exosomes. They observed that this peptide impaired MDSCs’ activity induced by cisplatin and 5-fluorouracil treatment, and potentiated the anti-tumor effect of these chemotherapeutic drugs
[30]. Additionally, Ono et al. showed that HSP90-EVs derived from metastatic oral cancer cells are taken up by macrophages, resulting in M2 polarization
[20].
Figure 1. The dual role of EV-HSPs inside the tumor microenvironment. (HSP: heat shock protein; MDSC: myeloid-derived suppressor cell; NK: natural killer). Created with BioRender.com (accessed on 10 May 2023).
On the contrary, there is also evidence that HSP-EVs can modulate innate immune responses, which leads to tumor control. Gastpar et al. demonstrated that high-HSP70/Bag-4 surface-positive exosomes act as natural killer (NK) cell attractants and elicit a strong NK lytic capacity to HSP70 membrane-positive tumors
[31]. Elsner et al. also found that HSP70-EVs derived from human melanoma cells induce the activation of mouse NK cells and result in tumor growth and metastasis reduction
[32]. Additionally, the encounter of myeloma-HSP-expressing exosomes and dendritic cells efficiently stimulates their maturation to promote T helper 1 (Th1) and cytotoxic T lymphocyte (CTL) anti-tumoral responses
[33]. Similarly, HSP70-enriched exosomes derived from a tumor heat-treatment promote tumor regression in murine models mediated by a Th1 immune response
[34]. Menay et al. showed the presence of HSP70 in the lumen and HSP90 on the surface of exosomes isolated from mice bearing a very aggressive T-cell lymphoma. The immunogenic properties of these HSP exosomes were found to induce Th1 response in naïve-syngeneic mice, resulting in protection against secondary challenges
[35]. Sen et al. demonstrated that the exposure of naïve murine macrophages is activated by HSP70-rich exosomes released from murine breast carcinoma cell lines post hyperthermia treatment. Moreover, other anti-tumoral responses were observed, such as increased macrophage migration and release of TNF-α and RANTES, which triggered a cytotoxic response against breast cancer cells
[36]. Vega et al. also showed that exosomes enriched in HSP70 activate macrophages to increase TNF-α production
[37].
Hurwitz MD et al. showed that the prostate cancer cell lines PC-3 and DU-145 secrete HSP72 exosomes after irradiation treatment. These exosomes also promote the increase in pro-inflammatory cytokines IL-6 and TNF-α and the expression of CD8+ T and NK cells
[38]. HSP-EVs play a pivotal role in stimulating an anti-tumor immune response after anticancer therapies. Lv et al. showed that hepatocarcinoma HepG2 cells secrete HSP-rich exosomes in response to paclitaxel, irinotecan, and carboplatin. These secreted EVs elicit an NK-cell-mediated anti-tumor response after granzyme B production. Exosome treatment in NK cells decreased the expression of inhibitory receptor CD94 and increased the expression of activating receptors CD69, NKG2D, and NKp44
[39].
All these reports demonstrate that the same HSP-EVs can sometimes act as a danger signal, increasing tumor immunogenicity and inducing an active response. On the other hand, HSP-expressing EVs can induce immunosuppression and compromise anticancer therapy efficacy. Furthermore, there is growing evidence that HSPs inside or on the membrane of EVs contribute to tumor progression and resistance to therapy. However, once the timeline and order of these events are understood, physicians can use them to manage the patient’s treatment better and improve their follow-ups (Table 1).
 +1 credit
+1 credit