Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mauro Maccarrone | -- | 3528 | 2024-01-17 10:33:54 | | | |
| 2 | Lindsay Dong | Meta information modification | 3528 | 2024-01-19 02:44:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tisi, A.; Palaniappan, S.; Maccarrone, M. Advanced Omics Techniques for Cochlear Research. Encyclopedia. Available online: https://encyclopedia.pub/entry/53960 (accessed on 27 July 2026).
Tisi A, Palaniappan S, Maccarrone M. Advanced Omics Techniques for Cochlear Research. Encyclopedia. Available at: https://encyclopedia.pub/entry/53960. Accessed July 27, 2026.
Tisi, Annamaria, Sakthimala Palaniappan, Mauro Maccarrone. "Advanced Omics Techniques for Cochlear Research" Encyclopedia, https://encyclopedia.pub/entry/53960 (accessed July 27, 2026).
Tisi, A., Palaniappan, S., & Maccarrone, M. (2024, January 17). Advanced Omics Techniques for Cochlear Research. In Encyclopedia. https://encyclopedia.pub/entry/53960
Tisi, Annamaria, et al. "Advanced Omics Techniques for Cochlear Research." Encyclopedia. Web. 17 January, 2024.
Copy Citation
Advanced genomics, transcriptomics, and epigenomics techniques are providing unprecedented insights into the understanding of the molecular underpinnings of the central nervous system, including the neuro-sensory cochlea of the inner ear.
omics
cochlea
single-cell omics
spatial omics
epigenomics
transcriptomics
genomics
1. Introduction
According to the World Health Organization (WHO), 432 million adults and 32 million children are affected by disabling hearing loss, and it is estimated that this number will increase to 700 million by 2050 [1]. In particular, sensorineural hearing loss (SNHL) is characterized by the deterioration of the neuro-sensory structure of the inner ear—the cochlea—and leads to irreversible hearing loss that affects communication, speech, and cognition, with a clear impact on the quality of life and severe socio-economic consequences. SNHL can be caused by either congenital or acquired factors (noise exposure, ototoxic drugs, ageing, strial or metabolic dysfunctions) [2]. The severity of the aetiology can range from synaptic disconnectivity of the sensory epithelium [3]—composed of inner (IHCs)/outer hair cells (OHCs) and supporting cells (SCs)—to critical cases of the loss of hair cells (HCs). The latter process is often followed by the degeneration of the downstream spiral ganglion neurons (SGNs) [4], whose axons form the auditory nerve. Although cochlear implants and hearing aids exhibit some beneficial outcomes in deaf patients, they cannot entirely replace the cochlea’s functionality [5]. Thus, management-based approaches must give way to disease-modifying interventions. This strategy needs a more thorough understanding of the molecular events that could eventually become novel therapeutic targets and/or diagnostic biomarkers of SNHL, to be exploited also in cochlear regeneration strategies. Thanks to the technological advancements in the field of molecular biology, recent progress has been made in identifying and characterizing novel genes involved in hearing loss [6], as well as new molecular mechanisms of cochlear development [7], degeneration, and regeneration [8].
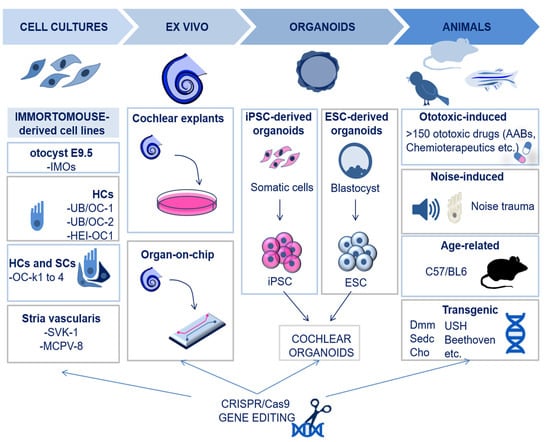
2. Experimental Models in Inner Ear Research
Modelling inner ear disorders is important to understand the molecular basis of hearing, as well as the mechanisms of deafness in humans. Currently, it is only possible to study human inner ear disorders in cadavers [9] since sampling tissues from alive subjects would cause irreversible damage to the intricate inner ear structures. Hence, this is possible only in cases of inner ear tumors [10][11]. Moreover, non-invasive techniques, such as magnetic resonance imaging (MRI) and computerized tomography (CT), cannot lead to a detailed understanding of the inner ear pathogenesis [12]. Therefore, most of the models for studies on the cochlea are based on cell cultures from animals or on animal models. Figure 1 summarizes the experimental models that are currently available and used in cochlear research.
Figure 1. Schematic illustration of the available experimental models for cochlear research. The available models for cochlear research include cell lines of otocyst, HCs, organ of Corti, and stria vascularis. Explants of cochlear tissues may also be used, more recently via microfluidic chambers for organ-on-chip cultures. Cochlear organoids are an additional in vitro possibility and can be derived from induced pluripotent stem cells (iPSCs) or from embryonic stem cells (ESCs). Animal models can be generated by exposure to ototoxic drugs or by noise trauma; also, age-related and transgenic models of hearing loss have been developed. Finally, all the models may be subjected to CRISPR/Cas9 to achieve targeted gene editing. Abbreviations: IMO; Immortomouse; HC; hair cell, SC; supporting cell, iPSC; induced pluripotent stem cell, ESC; embryonic stem cell, Dmm; disproportionate micromelia; sedc, spondyloepiphyseal dysplasia congenital; USH: Usher; Cho: chondrodysplasia.
3. Omics Techniques
3.1. Introduction to Omics: Principles and Advancements
The term omics refers to a rapidly evolving and expanding group of techniques aimed at investigating pools of biological molecules of an organism, including nucleic acids, proteins, and metabolites [13]. Hence, the main branches of omics techniques are known as genomics, epigenomics, transcriptomics, proteomics, and metabolomics. The most advanced omics techniques include single-cell omics and spatial omics, which allow the investigation of the molecular events occurring at a single-cell resolution and the retention of spatial information [14][15]. There are also other advanced and upcoming sequencing-based omics, such as epitranscriptomics, epiproteomics, and interactomics (DNA–RNA, RNA–RNA, RNA–protein, protein–protein, protein–metabolite), which give detailed information on the complex interactions and dynamics of regulation in a biological system [13]. The number of omics studies in cochlear research is relatively low compared to other sensory systems, since the sampling of the cochlear tissues has only recently advanced, and some techniques are incompatible with the small sample quantity obtained [16]. Nonetheless, the studies performed so far have significantly advanced the knowledge of cochlear physio-pathology.
3.2. Principles of Single-Cell Omics
The term single-cell omics refers to the process of profiling the genome, transcriptome, epigenome, proteome, and metabolome in individual cells. As a consequence, single-cell techniques were shown to be useful in several biological fields, including cancer [17], developmental biology [18], stem cell research [19], neuroscience [20], and hearing [8].
The first step of all these technologies is the isolation of individual cells and the setting up of libraries. Multiple methodologies have been designed to isolate single cells from pooled cell populations/tissues through a variety of techniques [21] that span from the most straightforward—using pipettes and cell isolation by dilution—to the more sophisticated—using advanced microfluidic technologies [22]. The latter include hydrodynamic trapping, droplet-based isolation, valve-based isolation, microwell-based isolation and dielectrophoresis trapping [23], as well as magnetic-activated cell sorting (MACS), flow-activated cell sorting (FACS) [21], laser capture microdissection (LCM)—which also preserves spatial context—and nanowell-based cell sorting [24]. Details of the isolation methods for abundant or rare cells have been described by Wang and Navin [25].
After the isolation of single cells, the genome, the epigenome and the transcriptome can be profiled [8][26]. Notably, single-cell multi-omics approaches have recently been developed to investigate the molecular events that occur in individual cells under physiological or pathological conditions in a wider overview, at once. An example of this cutting-edge methodology is single-cell triple-omics sequencing (scTrio-seq), which simultaneously gathers data from the genome, DNA methylome, and transcriptome of a single cell [27].
3.3. Spatial Omics
The study of omics at a single-cell resolution has been transformative in the identification of novel biomarkers and molecular regulators of tissues, yet single-cell omics cannot deliver information on the tissue or sub-cellular localization of the isolated cells. For this reason, spatial omics have been developed with the aim of identifying molecular events while maintaining the spatial information. Multiple spatial omics approaches exist, and they vary depending on the biomolecules of interest. In the cochlea, spatial omics are of particular relevance due to its complex anatomical architecture. Indeed, the cochlea exhibits a tonotopic organization from its base (high-frequency perception) to the apex (low-frequency perception), which requires appropriate cellular structures and expression patterns [28]. Moreover, different cell types are also present from the medial (i.e., greater epithelial ridge (GER), IHCs, and their associated SCs) to the lateral (i.e., Deiters’ cells, pillar cells, and OHCs) compartment of the cochlea [28].
4. The Role of Bioinformatics in Analyzing Omics Data
In order to fill the knowledge gap between omics data acquisition and interpretation, bioinformatics is a critical field. Numerous computational techniques have been developed to this end, including machine learning, deep learning, data mining and statistical and metaheuristic approaches, to analyze, process, interpret, and integrate omics data for both single omics and integrative multi-omics [29][30][31][32][33]. Machine learning and deep learning are frequently used in the research community for decoding and analyzing data, predicting disease occurrence and recurrence, calculating survival rates, and finding potential biomarkers [29]. Deep learning models are a sub-set of machine learning tools of high utility since they are automated and analyze large high-dimensional data sets. Deep learning is primarily based on stratified artificial neural networks, providing diverse interpretations based on the fed data. The primary neural networks in deep learning include recursive neural networks (RvNNs), recurrent neural networks (RNNs), and convolutional neural networks (CNNs) [34]. Given the recent rapid advancements in omics and the accumulation of high-throughput omics data, future efforts should be aimed towards improving current machine learning and deep learning models for multi-omic data analysis. In this context, graph neural networks (GNNs) have gained attention in recent years [35]: the spatial relations within and between cells can be better represented with graph models, and graph-based artificial intelligence appears to hold promise, especially with regard to the most advanced omics (i.e, spatial omics). In this context, it should be mentioned that two relevant techniques have recently been developed to analyze spatial transcriptomics data: SPAcI [36] and SiGra [37]. They both have several technical advantages over existing methods, such as an improvement in accuracy, enhancing noisy gene expression data sets, and an increased ability to adapt [35][36].
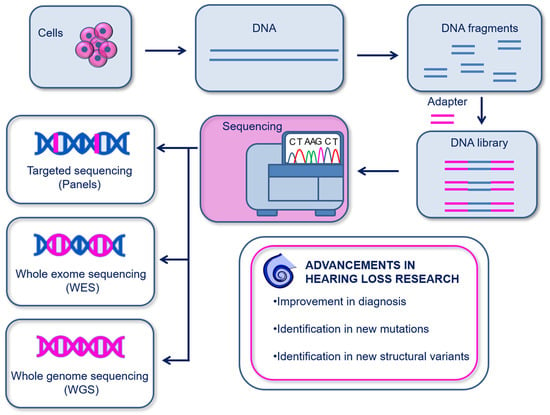
5. Genomics
5.1. Principles of Sequencing
Genomics investigates somatic and germ-line inter-individual variations in the genome. The currently most used genomics are based on sequencing for the determination of the nucleic acid sequence. Genomics has been used to identify several genetic disorders and to disclose novel alleles in multiple inherited human diseases [13], including hearing loss [38][39][40][41][42][43]. The first sequencing method, known as the chain-termination method, was first developed by Sanger in 1977 and was based on the capillary electrophoresis of fragmented DNA bound to a single-stranded DNA template. The main drawback of Sanger sequencing is the ability to sequence only a low amount of DNA at a time [44]. To date, more advanced sequencing technologies have been developed and allow massive, faster, and more precise sequencing of nucleic acids. These are next generation sequencing (NGS)—more widely used—and third generation sequencing (TGS). The primary difference between these two techniques is the DNA read length. In NGS, the DNA is cleaved in small fragments (150–1000 bp), then amplified and sequenced; instead, TGS uses single-molecule sequencing without the need for prior amplification and reads long DNA sequences at a time. Moreover, it is possible to sequence different lengths of the genome depending on the experimental purpose: targeted genes (targeted panel sequencing), whole-exome sequencing (WES) [40][41] or whole-genome sequencing (WGS) [42][43] (Figure 2).
Figure 2. Schematic illustration of genomics. DNA is isolated from cells or tissues and is fragmented in order to create DNA libraries using DNA adapters. Sequencing can then be performed on targeted sequences (panels), on the whole exosome (WES), or on the whole genome (WGS). Genomics has provided important advancements in the diagnosis and discovery of genetic hearing loss.
5.2. Single-Cell and Spatial Genomics
Single-cell DNA sequencing (scDNAseq) allows the DNA profiling of individual cells [45] and is generally based on NGS. The whole genome of single cells can be primarily amplified using three methods: (i) the degenerate oligonucleotide-primed PCR (DOP-PCR), (ii) the multiple displacement amplification (MDA), and (iii) the multiple annealing and looping-based amplification cycles (MALBAC) [46]. Recently, a single-cell WGS method based on TGS was also developed in order to sequence long reads; this is known as “single-molecule real-time sequencing of long fragments amplified through transposon insertion” (SMOOTH-seq) [46]. SMOOTH-seq has greatly improved the identification of structural variants (SVs) and extra-chromosomal DNA compared to NGS [47].
5.3. Genomic Studies Have Delivered Unprecedented Knowledge on the Genetic Background and Early Diagnosis of Inherited Hearing Loss
Genetic hearing loss affects any part of the auditory system and accounts for ~50% of the deaf population. It can be either non-syndromic (70%) [48][49] or syndromic (30%) [50]. The large heterogeneity of genes involved in deafness makes it difficult to study and diagnose it [40]. However, thanks to the advancements in genomics, to date several variants have been identified in genes associated with hearing loss. For instance, the combination of WES, qPCR, and TGS was able to unravel for the first-time novel SVs of centrosomal protein 78 (CEP78), a key gene responsible for hearing loss associated with cone–rod dystrophy (CRDHL) [43]. The applications of advanced genomics have also revealed new variants that have recently been outlined in important hearing loss-related genes, namely, myosin 15 A (MYO15A), otoferlin (OTOF), radixin (RDX) [51], TATA-box-binding protein-associated factor 1 (TAF1) [52], atonal BHLH transcription factor 1 (ATOH1) [53], and centrosomal protein 78 (CEP78) [43]. The discovery of novel variants represents a fundamental step forward in the understanding of the molecular basis of cochlear diseases, and indeed, it has improved the diagnosis of genetic hearing loss, as well as the prediction of its severity and prognosis.
6. Transcriptomics
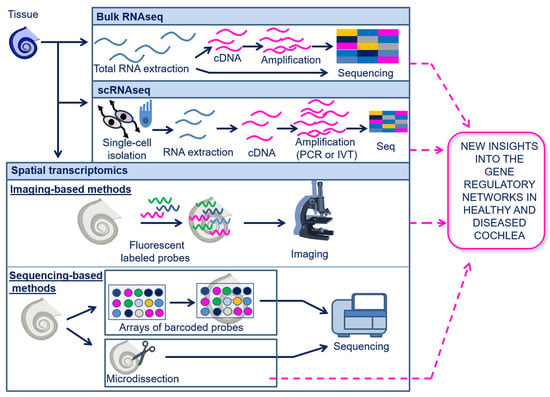
Transcriptomics enable the analysis of gene expression at the RNA level, including messenger RNAs (mRNAs), transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), and other non-coding RNAs (ncRNAs) (e.g., microRNAs (miRNAs), long-non-coding RNAs (lncRNAs), and circular RNAs (circRNAs)) [13][54]. As for genomics, the currently most used transcriptomics technologies are based on sequencing (described above in Section 5.1); however, transcriptome profiling is more challenging compared to genome sequencing due to the highly dynamic nature of the transcriptome in biological processes. The sequencing of the entire transcriptome in a tissue or cell population is known as bulk-RNAseq and can be performed either with direct RNA sequencing (dRNA-seq) or with cDNA sequencing [55]. A schematic overview of the transcriptomics techniques is shown in Figure 3.
Figure 3. Schematic illustration of transcriptomics. Transcriptomics can be performed at a bulk, single-cell, or spatial resolution. In bulk RNAseq, total RNA is extracted from the tissue and can be directly sequenced or converted into cDNA and then sequenced. In scRNA-seq, the sequencing of the RNA is limited to single cells that are isolated from the tissue and analyzed individually. In spatial transcriptomics, the transcriptome may be analyzed with imaging-based methods, using fluorescent labeled probes which bind to the RNA on tissue slides, followed by microscopic analysis; spatial transcriptomics may also be performed through sequencing-based methods using arrays of barcoded probes or microdissection of target tissue areas, both followed by sequencing. Array-based spatial transcriptomics have not yet been applied in cochlear research. The other transcriptomics techniques have provided important new insights into the gene regulatory networks of the cochlea, under both physiological and pathological conditions. Abbreviations: Bulk RNA seq, Bulk RNA sequencing; scRNA-seq, single-cell RNA sequencing.
7. Epigenomics
7.1. Principles of Epigenomics
The term epigenomics refers to the techniques used to investigate the epigenome, which is the set of regulatory processes that modify the activity of gene expression without modifications in the DNA sequence. Epigenomics can be classified depending on the target: DNA methylation, histone modifications, chromatin accessibility, and chromosome interactions. The methodologies to study bulk epigenomics can be further classified as “array-based” and “sequencing-based” techniques [56]. Array-based technologies use hybridization with pre-designed microarrays, while sequencing-based methods use techniques like NGS. DNA methylation is an epigenetic mark where methyl groups are added to the cytosine bases of the DNA. It is important to highlight that to investigate DNA methylation, a required first step is the exposure of the methylated DNA through one of the following methods: (i) DNA digestion by methylation-sensitive restriction enzymes (MSREs) [57], (ii) affinity enrichment of DNA by antibodies targeting methylated CpGs [58], or (iii) conversion of unmethylated cytosines to uracil by bisulfite treatment [57]. To date, the bisulfite sequencing (BS-seq) method is considered the gold-standard technique for studies on DNA methylation because of its single-base resolution [56]. Histones can be modified primarily through acetylation, phosphorylation, methylation, and other miscellaneous modifications [59]. One of the most used techniques for monitoring histone modifications is chromatin immunoprecipitation (ChIP), in which the histone modifications of interest are targeted by antibodies. The cleavage under targets and release using nuclease (CUT&RUN) [60] and cleavage under targets and tagmentation (CUT&TAG) [61] methods are additional techniques used for the analysis of histone modifications; both rely on the same principle of recognizing DNA-bound proteins of interest through specific antibodies. The chromatin is highly dynamic, allowing regulators (enhancers, promoters, and chromatin-binding factors, among others) to have multiple physical interactions with DNA, thereby playing an important role in regulating gene expression. Multiple techniques for chromatin accessibility studies have also been developed. Among these, the most recent is the accessible chromatin using sequencing technology (ATAC-seq). It employs tagmentation (inserting adapter sequences by using the hyperactive mutant Tn5 transposase) to open target regions of the chromatin, which are then amplified and sequenced [62]. Other widely used techniques for chromatin accessibility include the DNase I hyper-sensitive sites sequencing (DNAse-seq) [63], the micrococcal nuclease digestion with deep sequencing (MNase-seq) [64], and the formaldehyde-assisted identification of regulatory elements followed by sequencing (FAIRE-seq) [65]. The higher-order organization of the nucleus is also important for the epigenetic regulation of cellular processes; hence, techniques able to analyze chromosomal interactions have also been developed. They include the chromatin conformation capture technique (3C), Hi-C, the chromatin interaction analysis by paired-end tag sequencing (ChIA PET), and the proximity ligation-assisted ChIP-seq (PLAC-seq) [56].
7.2. Single-Cell Epigenomics
Single-cell epigenomics enable a detailed analysis of the epigenetic regulation at a single-cell resolution, which includes single-cell DNA methylation profiling, single-cell chromatin mapping, single-cell Hi-C, and single-cell replication dynamics [66]. Several methods for single-cell DNA methylation profiling exist, the most recent of which are “single-cell combinatorial indexing for methylation analysis” (sci-MET) and “single-cell CGI methylation sequencing” (scCGI-seq) [56][67][68]. Histone modifications in single cells can also be studied using sc-ChIP-seq, single-cell droplet-based chromatin immunoprecipitation (drop-ChIP) [26], single-cell chromatin immune-cleavage sequencing technique (scChIC-seq), antibody-guided chromatin tagmentation sequencing (ACT-seq), combinatorial barcoding and targeted chromatin release (COBATCH), and single-cell chromatin integration labeling sequencing (scChIL-seq) [56]. Finally, single-cell chromatin accessibility can be investigated using scDNAse-seq and scATAC-seq. The available single-cell epigenetic methods have been recently reviewed in detail [66][69].
7.3. Spatial Epigenomics
To fully appreciate the influence of epigenetic variations in patho-physiological processes, it is essential to know their spatial context. However, the development of spatial epigenomics techniques has been challenging for a long time due to the limited spatial resolution available [70][71]. The first spatial epigenomic technology was developed in 2021 and is now beginning to open new possibilities in the field of biology and medicine. The first spatial epigenomic technique that has been developed is the “high-spatial-resolution chromatin modification state profiling by sequencing” (hsrChST-seq). It is based on the spatial transcriptomic technique DBiT-seq, in which there is a combination of CUT&TAG and tissue deterministic barcoding with fluorescence microscopy [72]. Another technique developed later on to resolve chromatin accessibility spatially is the spatial-ATAC seq, which is based on the combination of in situ Tn5 transposase chemistry with microfluidic deterministic barcoding [73]. Recently, it has been possible to analyze the active and inactive promoters/enhancers associated with histone modifications in single cells while maintaining spatial information thanks to the advent of epigenomic MERFISH [73]. Epigenomic MERFISH combines CUT&TAG and MERFISH (a spatial epigenomic technique for the analysis of histone modifications) [74].
7.4. Epigenetic Profiling of the Cochlea Has Provided New Insights into the Mechanisms Whereby Genes Responsible for Auditory Function Are Regulated
Hearing loss can be caused by epigenetic alterations or by mutations in the genes encoding for the epigenetic machinery, affecting DNA methylation dynamics [75][76][77], histone modifications [78][79][80], and chromatin remodeling [76][81][82]. Thus, investigating epigenetic mechanisms could eventually pave the way towards new approaches to therapeutics. To date, most of the studies on the cochlear epigenome are based on bulk epigenomic profiling, and only a few were performed with single-cell epigenomics, namely scATAC-seq [83][84]. Instead, spatial epigenomics has not yet been applied in this field, though the epigenomics studies conducted until now have given us profound insights into the regulatory mechanisms of development, trans-differentiation, and regeneration of the auditory system. The application of ChIP-seq and ChIP-qPCR has led to the identification of fundamental epigenetic modifications in the promoters of two key genes involved in SGN differentiation (Cdk2 and NeuroD1), which affects the binding of the regulatory transcription factor neurogenin 1 (neurog1) [85]. Also, ChIP-qPCR allowed for the description of the histone modifications associated with the epigenetic regulation of atonal bHLH transcription factor 1 (Atoh1), which is an evolutionarily conserved transcription factor for the development of the auditory system [86]. Yet, histone modifications of Atoh1, which are characteristic of HCs during their development, are suppressed in the same cells after birth, but they persist in perinatal SCs. This is an important finding, which gives new information on the mechanisms underlying the regenerative potential of SCs [86].
8. Conclusions
Overall, advanced genomics, epigenomics, and transcriptomics techniques represent the state-of-the-art approaches in cochlear research and are providing unprecedented knowledge on the molecular basis of cochlear patho-physiology. There are various open questions in the field of cochlear research that could be addressed through the application of omic techniques. For instance, in the context of the heterogeneity of the cochlear tissue, as of now, eight different SC populations with distinct morphologies have been identified, but little is known about their contribution to auditory function [87]. The application of single-cell and spatial transcriptomic technologies could provide a map of cell-type-specific gene expression patterns that can help to hypothesize the potential role of these cells in auditory processes. In addition, the application of single-cell epigenomics could enhance our understanding of the important cell-type-specific gene regulatory networks that can broaden our knowledge on the putative role of SC in cochlear homeostasis. Another active area of research in hearing is the mechanisms underlying ototoxicity. For instance, cisplatin, one of the primary chemotherapeutic agents utilized in oncology, has debilitating effects on hearing function and, hence, quality of life. Although efforts have been undertaken to understand the molecular underpinnings behind cisplatin-induced ototoxicity, to date, no effective therapeutics have been approved to counteract such adverse effects [88]. This necessitates a thorough molecular comprehension of the ototoxic processes from cisplatin trafficking to downstream signaling. The application of single-cell and spatial omics can give insights into the consequences of those ototoxic compounds. For instance, genomics would provide advanced knowledge on the cell-type-specific mutations and their location, transcriptomics on the cell-type-specific biomarkers expressed under ototoxic conditions, and epigenomics on putative gene regulatory dynamics in ototoxicity, which can all provide novel pharmacological/gene therapy targets not only for HCs (where most of the research is focused on [89]) but also for SCs, which are known for their role in ototoxicity [90].
References
- Deafness and Hearing Loss. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 23 August 2023).
- Ma, Y.; Wise, A.K.; Shepherd, R.K.; Richardson, R.T. New Molecular Therapies for the Treatment of Hearing Loss. Pharmacol. Ther. 2019, 200, 190–209.
- Liberman, M.C.; Kujawa, S.G. Cochlear Synaptopathy in Acquired Sensorineural Hearing Loss: Manifestations and Mechanisms. Hear. Res. 2017, 349, 138–147.
- Wise, A.K.; Pujol, R.; Landry, T.G.; Fallon, J.B.; Shepherd, R.K. Structural and Ultrastructural Changes to Type I Spiral Ganglion Neurons and Schwann Cells in the Deafened Guinea Pig Cochlea. J. Assoc. Res. Otolaryngol. 2017, 18, 751.
- Smith-Cortinez, N.; Tan, A.K.; Stokroos, R.J.; Versnel, H.; Straatman, L.V. Regeneration of Hair Cells from Endogenous Otic Progenitors in the Adult Mammalian Cochlea: Understanding Its Origins and Future Directions. Int. J. Mol. Sci. 2023, 24, 7840.
- Santos-Cortez, R.L.P.; Yarza, T.K.L.; Bootpetch, T.C.; Tantoco, M.L.C.; Mohlke, K.L.; Cruz, T.L.G.; Perez, M.E.C.; Chan, A.L.; Lee, N.R.; Tobias-Grasso, C.A.M.; et al. Identification of Novel Candidate Genes and Variants for Hearing Loss and Temporal Bone Anomalies. Genes 2021, 12, 566.
- Kelley, M.W. Cochlear Development; New Tools and Approaches. Front. Cell Dev. Biol. 2022, 10, 884240.
- Wu, M.; Xia, M.; Li, W.; Li, H. Single-Cell Sequencing Applications in the Inner Ear. Front. Cell Dev. Biol. 2021, 9, 637779.
- Bommakanti, K.; Iyer, J.S.; Stankovic, K.M. Cochlear Histopathology in Human Genetic Hearing Loss: State of the Science and Future Prospects. Hear. Res. 2019, 382, 107785.
- Yamakami, I.; Ito, S.; Higuchi, Y. Retrosigmoid Removal of Small Acoustic Neuroma: Curative Tumor Removal with Preservation of Function: Clinical Article. J. Neurosurg. 2014, 121, 554–563.
- Nicoli, T.K.; Atula, T.; Sinkkonen, S.T.; Korpi, J.; Vnencak, M.; Tarkkanen, J.; Mäkitie, A.A.; Jero, J. Ear Canal and Middle-Ear Tumors: A Single-Institution Series of 87 Patients. Acta Oto-Laryngol. 2022, 142, 132–139.
- Gao, S.S.; Xia, A.; Applegate, B.E.; Shelton, R.L.; Yuan, T.; Raphael, P.D.; Oghalai, J.S. Quantitative Imaging of Cochlear Soft Tissues in Wild-Type and Hearing-Impaired Transgenic Mice by Spectral Domain Optical Coherence Tomography. Opt. Express 2011, 19, 15415–15428.
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861.
- Kong, S.; Li, R.; Tian, Y.; Zhang, Y.; Lu, Y.; Ou, Q.; Gao, P.; Li, K.; Zhang, Y. Single-Cell Omics: A New Direction for Functional Genetic Research in Human Diseases and Animal Models. Front. Genet. 2023, 13, 1100016.
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-Omics: Novel Approaches to Probe Cell Heterogeneity and Extracellular Matrix Biology. Matrix Biol. 2020, 91–92, 152–166.
- Zheng, Q.Y.; Rozanas, C.R.; Thalmann, I.; Chance, M.R.; Alagramam, K.N. Inner Ear Proteomics of Mouse Models for Deafness, a Discovery Strategy. Brain Res. 2006, 1091, 113–121.
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–95.
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-Coverage Single-Cell MRNA Sequencing Reveals Cellular Heterogeneity and Activated Signaling Pathways in Developing Cerebral Cortex. Nat. Biotechnol. 2014, 32, 1053–1058.
- Ealy, M.; Ellwanger, D.C.; Kosaric, N.; Stapper, A.P.; Heller, S. Single-Cell Analysis Delineates a Trajectory toward the Human Early Otic Lineage. Proc. Natl. Acad. Sci. USA 2016, 113, 8508–8513.
- Tasic, B. Single Cell Transcriptomics in Neuroscience: Cell Classification and Beyond. Curr. Opin. Neurobiol. 2018, 50, 242–249.
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017, 33, 155–168.
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018, 50, 1–14.
- Xu, X.; Wang, J.; Wu, L.; Guo, J.; Song, Y.; Tian, T.; Wang, W.; Zhu, Z.; Yang, C. Microfluidic Single-Cell Omics Analysis. Small 2020, 16, 1903905.
- Menze, L.; Duarte, P.A.; Haddon, L.; Chu, M.; Chen, J. Selective Single-Cell Sorting Using a Multisectorial Electroactive Nanowell Platform. ACS Nano 2022, 16, 211–220.
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609.
- Mincarelli, L.; Lister, A.; Lipscombe, J.; Macaulay, I.C. Defining Cell Identity with Single-Cell Omics. Proteomics 2018, 18, 1700312.
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-Cell Triple Omics Sequencing Reveals Genetic, Epigenetic, and Transcriptomic Heterogeneity in Hepatocellular Carcinomas. Cell Res. 2016, 26, 304–319.
- Raphael, Y.; Altschuler, R.A. Structure and Innervation of the Cochlea. Brain Res. Bull. 2003, 60, 397–422.
- Kaur, P.; Singh, A.; Chana, I. Computational Techniques and Tools for Omics Data Analysis: State-of-the-Art, Challenges, and Future Directions. Arch. Comput. Methods Eng. 2021, 28, 4595–4631.
- Hie, B.; Peters, J.; Nyquist, S.K.; Shalek, A.K.; Berger, B.; Bryson, B.D. Computational Methods for Single-Cell RNA Sequencing. Annu. Rev. Biomed. Data Sci. 2020, 3, 339–364.
- Stanojevic, S.; Li, Y.; Ristivojevic, A.; Garmire, L.X. Computational Methods for Single-Cell Multi-Omics Integration and Alignment. Genom. Proteom. Bioinform. 2022, 20, 836–849.
- Li, Y.; Stanojevic, S.; Garmire, L.X. Emerging Artificial Intelligence Applications in Spatial Transcriptomics Analysis. Comput. Struct. Biotechnol. J. 2022, 20, 2895–2908.
- Efremova, M.; Teichmann, S.A. Computational Methods for Single-Cell Omics across Modalities. Nat. Methods 2020, 17, 14–17.
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of Deep Learning: Concepts, CNN Architectures, Challenges, Applications, Future Directions. J. Big Data 2021, 8, 53.
- Wang, H.; Guo, F.; Du, M.; Wang, G.; Cao, C. A Novel Method for Drug-Target Interaction Prediction Based on Graph Transformers Model. BMC Bioinform. 2022, 23, 459.
- Tang, Z.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SpaCI: Deciphering Spatial Cellular Communications through Adaptive Graph Model. Brief. Bioinform. 2023, 24, bbac563.
- Tang, Z.; Li, Z.; Hou, T.; Zhang, T.; Yang, B.; Su, J.; Song, Q. SiGra: Single-Cell Spatial Elucidation through an Image-Augmented Graph Transformer. Nat. Commun. 2023, 14, 5618.
- Rehman, A.U.; Morell, R.J.; Belyantseva, I.A.; Khan, S.Y.; Boger, E.T.; Shahzad, M.; Ahmed, Z.M.; Riazuddin, S.; Khan, S.N.; Riazuddin, S.; et al. Targeted Capture and Next-Generation Sequencing Identifies C9orf75, Encoding Taperin, as the Mutated Gene in Nonsyndromic Deafness DFNB79. Am. J. Hum. Genet. 2010, 86, 378–388.
- Yan, D.; Tekin, M.; Blanton, S.H.; Liu, X.Z. Next-Generation Sequencing in Genetic Hearing Loss. Genet. Test. Mol. Biomark. 2013, 17, 581–587.
- Shearer, A.E.; Smith, R.J.H. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss. Otolaryngol.–Head Neck Surg. 2015, 153, 175–182.
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9, 314696.
- Hegde, M.; Santani, A.; Mao, R.; Ferreira-Gonzalez, A.; Weck, K.E.; Voelkerding, K.V. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch. Pathol. Lab. Med. 2017, 141, 798–805.
- Ascari, G.; Rendtorff, N.D.; De Bruyne, M.; De Zaeytijd, J.; Van Lint, M.; Bauwens, M.; Van Heetvelde, M.; Arno, G.; Jacob, J.; Creytens, D.; et al. Long-Read Sequencing to Unravel Complex Structural Variants of CEP78 Leading to Cone-Rod Dystrophy and Hearing Loss. Front. Cell Dev. Biol. 2021, 9, 664317.
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467.
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of Single-Cell DNA Sequencing. Annu. Rev. Genom. Hum. Genet. 2021, 22, 171.
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 2016, 17, 175–188.
- Fan, X.; Yang, C.; Li, W.; Bai, X.; Zhou, X.; Xie, H.; Wen, L.; Tang, F. SMOOTH-Seq: Single-Cell Genome Sequencing of Human Cells on a Third-Generation Sequencing Platform. Genome Biol. 2021, 22, 195.
- Zhong, L.X.; Kun, S.; Jing, Q.; Jing, C.; Denise, Y. Non-Syndromic Hearing Loss and High-Throughput Strategies to Decipher Its Genetic Heterogeneity. J. Otol. 2013, 8, 6–24.
- Aldè, M.; Cantarella, G.; Zanetti, D.; Pignataro, L.; Mantia, I.L.; Maiolino, L.; Ferlito, S.; Mauro, P. Di; Cocuzza, S.; Lechien, J.R.; et al. Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review. Biomedicines 2023, 11, 1616.
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss—Syndromic. Otolaryngol. Clin. N. Am. 2015, 48, 1041.
- Bai, X.; Nian, S.; Feng, L.; Ruan, Q.; Luo, X.; Wu, M.; Yan, Z. Identification of Novel Variants in MYO15A, OTOF, and RDX with Hearing Loss by next-Generation Sequencing. Mol. Genet. Genom. Med. 2019, 7, e808.
- Cheng, H.; Capponi, S.; Wakeling, E.; Marchi, E.; Li, Q.; Zhao, M.; Weng, C.; Stefan, P.G.; Ahlfors, H.; Kleyner, R.; et al. Missense Variants in TAF1 and Developmental Phenotypes: Challenges of Determining Pathogenicity. Hum. Mutat. 2020, 41, 449–464.
- Brownstein, Z.; Gulsuner, S.; Walsh, T.; Martins, F.T.A.; Taiber, S.; Isakov, O.; Lee, M.K.; Bordeynik-Cohen, M.; Birkan, M.; Chang, W.; et al. Spectrum of Genes for Inherited Hearing Loss in the Israeli Jewish Population, Including the Novel Human Deafness Gene ATOH1. Clin. Genet. 2020, 98, 353–364.
- Dong, Z.C.; Chen, Y. Transcriptomics: Advances and Approaches. Sci. China Life Sci. 2013, 56, 960–967.
- Wongsurawat, T.; Jenjaroenpun, P.; Wanchai, V.; Nookaew, I. Native RNA or CDNA Sequencing for Transcriptomic Analysis: A Case Study on Saccharomyces Cerevisiae. Front. Bioeng. Biotechnol. 2022, 10, 842299.
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A Comparative Overview of Epigenomic Profiling Methods. Front. Cell Dev. Biol. 2021, 9, 714687.
- Oakes, C.C.; La Salle, S.; Robaire, B.; Trasler, J.M. Evaluation of a Quantitative DNA Methylation Analysis Technique Using Methylation-Sensitive/Dependent Restriction Enzymes and Real-Time PCR. Epigenetics 2006, 1, 146–152.
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-Wide and Promoter-Specific Analyses Identify Sites of Differential DNA Methylation in Normal and Transformed Human Cells. Nat. Genet. 2005, 37, 853–862.
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395.
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. eLife 2017, 6, e21856.
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nat. Commun. 2019, 10, 1930.
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9.
- Song, L.; Crawford, G.E. DNase-Seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384.
- Pajoro, A.; Muiño, J.M.; Angenent, G.C.; Kaufmann, K. Profiling Nucleosome Occupancy by MNase-Seq: Experimental Protocol and Computational Analysis. Methods Mol. Biol. 2018, 1675, 167–181.
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-Seq and FAIRE-Seq Open Chromatin Profiling. PLoS Genet. 2015, 11, e1004994.
- Schwartzman, O.; Tanay, A. Single-Cell Epigenomics: Techniques and Emerging Applications. Nat. Rev. Genet. 2015, 16, 716–726.
- Han, L.; Wu, H.J.; Zhu, H.; Kim, K.Y.; Marjani, S.L.; Riester, M.; Euskirchen, G.; Zi, X.; Yang, J.; Han, J.; et al. Bisulfite-Independent Analysis of CpG Island Methylation Enables Genome-Scale Stratification of Single Cells. Nucleic Acids Res. 2017, 45, e77.
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly Scalable Generation of DNA Methylation Profiles in Single Cells. Nat. Biotechnol. 2018, 36, 428–431.
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-Cell Epigenomics: Powerful New Methods for Understanding Gene Regulation and Cell Identity. Genome Biol. 2016, 17, 72.
- Su, J.H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659.e26.
- Takei, Y.; Yun, J.; Zheng, S.; Ollikainen, N.; Pierson, N.; White, J.; Shah, S.; Thomassie, J.; Suo, S.; Eng, C.H.L.; et al. Integrated Spatial Genomics Reveals Global Architecture of Single Nuclei. Nature 2021, 590, 344–350.
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially Resolved Chromatin Modification Profiling at the Cellular Level. Science 2022, 375, 681–686.
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial Profiling of Chromatin Accessibility in Mouse and Human Tissues. Nature 2022, 609, 375–383.
- Lu, T.; Ang, C.E.; Zhuang, X. Spatially Resolved Epigenomic Profiling of Single Cells in Complex Tissues. Cell 2022, 185, 4448–4464.e17.
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 Mutation Hot Spot Causes Varied Phenotypes of HSAN1 with Dementia and Hearing Loss. Neurology 2013, 80, 824–828.
- Balendran, V.; Ritter, K.E.; Martin, D.M. Epigenetic Mechanisms of Inner Ear Development. Hear. Res. 2022, 426, 108440.
- Seyama, R.; Tsuchida, N.; Okada, Y.; Sakata, S.; Hamada, K.; Azuma, Y.; Hamanaka, K.; Fujita, A.; Koshimizu, E.; Miyatake, S.; et al. Two Families with TET3-Related Disorder Showing Neurodevelopmental Delay with Craniofacial Dysmorphisms. J. Hum. Genet. 2022, 67, 157–164.
- Ahmed, M.; Streit, A. Lsd1 Interacts with CMyb to Demethylate Repressive Histone Marks and Maintain Inner Ear Progenitor Identity. Development 2018, 145, dev160325.
- Shin, J.O.; Lee, J.J.; Kim, M.; Chung, Y.W.; Min, H.; Kim, J.Y.; Kim, H.P.; Bok, J. CTCF Regulates Otic Neurogenesis via Histone Modification in the Neurog1 Locus. Moleucles Cells 2018, 41, 695–702.
- Tao, L.; Yu, H.V.; Llamas, J.; Trecek, T.; Wang, X.; Stojanova, Z.; Groves, A.K.; Segil, N. Enhancer Decommissioning Imposes an Epigenetic Barrier to Sensory Hair Cell Regeneration. Dev. Cell 2021, 56, 2471–2485.e5.
- Vissers, L.E.L.M.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome. Nat. Genet. 2004, 36, 955–957.
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of Chromatin Remodeling Gene Cecr2 in Neurulation and Inner Ear Development. Dev. Dyn. 2011, 240, 372–383.
- Iyer, A.A.; Hosamani, I.; Nguyen, J.D.; Cai, T.; Singh, S.; McGovern, M.M.; Beyer, L.; Zhang, H.; Jen, H.I.; Yousaf, R.; et al. Cellular Reprogramming with ATOH1, GFI1, and POU4F3 Implicate Epigenetic Changes and Cell-Cell Signaling as Obstacles to Hair Cell Regeneration in Mature Mammals. eLife 2022, 11, e79712.
- Jimenez, E.; Slevin, C.C.; Song, W.; Chen, Z.; Frederickson, S.C.; Gildea, D.; Wu, W.; Elkahloun, A.G.; Ovcharenko, I.; Burgess, S.M. A Regulatory Network of Sox and Six Transcription Factors Initiate a Cell Fate Transformation during Hearing Regeneration in Adult Zebrafish. Cell Genom. 2022, 2, 100170.
- Song, Z.; Jadali, A.; Fritzsch, B.; Kwan, K.Y. NEUROG1 Regulates CDK2 to Promote Proliferation in Otic Progenitors. Stem Cell Rep. 2017, 9, 1516–1529.
- Fu, Y.; Yuan, S.S.; Zhang, L.J.; Ji, Z.L.; Quan, X.J. Atonal BHLH Transcription Factor 1 Is an Important Factor for Maintaining the Balance of Cell Proliferation and Differentiation in Tumorigenesis (Review). Oncol. Lett. 2020, 20, 2595–2605.
- McGovern, M.M.; Randle, M.R.; Cuppini, C.L.; Graves, K.A.; Cox, B.C. Multiple Supporting Cell Subtypes Are Capable of Spontaneous Hair Cell Regeneration in the Neonatal Mouse Cochlea. Development 2019, 146, dev171009.
- Wang, X.; Zhou, Y.; Wang, D.; Wang, Y.; Zhou, Z.; Ma, X.; Liu, X.; Dong, Y. Cisplatin-Induced Ototoxicity: From Signaling Network to Therapeutic Targets. Biomed. Pharmacother. 2023, 157, 114045.
- Wang, J.; Zheng, J.; Wang, H.; He, H.; Li, S.; Zhang, Y.; Wang, Y.; Xu, X.; Wang, S. Gene Therapy: An Emerging Therapy for Hair Cells Regeneration in the Cochlea. Front. Neurosci. 2023, 17, 1177791.
- Waissbluth, S.; Maass, J.C.; Sanchez, H.A.; Martínez, A.D. Supporting Cells and Their Potential Roles in Cisplatin-Induced Ototoxicity. Front. Neurosci. 2022, 16, 867034.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
436
Revisions:
2 times
(View History)
Update Date:
19 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No