Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ana Robles-Martín | -- | 1617 | 2024-01-10 12:24:17 | | | |
| 2 | Jessie Wu | -1 word(s) | 1616 | 2024-01-11 03:46:54 | | | | |
| 3 | Jessie Wu | Meta information modification | 1616 | 2024-01-12 08:54:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Robles-Martín, A.; Roda, S.; Muñoz-Tafalla, R.; Guallar, V. Develop a PluriZyme. Encyclopedia. Available online: https://encyclopedia.pub/entry/53681 (accessed on 29 June 2026).
Robles-Martín A, Roda S, Muñoz-Tafalla R, Guallar V. Develop a PluriZyme. Encyclopedia. Available at: https://encyclopedia.pub/entry/53681. Accessed June 29, 2026.
Robles-Martín, Ana, Sergi Roda, Rubén Muñoz-Tafalla, Victor Guallar. "Develop a PluriZyme" Encyclopedia, https://encyclopedia.pub/entry/53681 (accessed June 29, 2026).
Robles-Martín, A., Roda, S., Muñoz-Tafalla, R., & Guallar, V. (2024, January 10). Develop a PluriZyme. In Encyclopedia. https://encyclopedia.pub/entry/53681
Robles-Martín, Ana, et al. "Develop a PluriZyme." Encyclopedia. Web. 10 January, 2024.
Copy Citation
Protein engineering is the design and modification of protein structures to optimize their functions or create novel functionalities for applications in biotechnology, medicine or industry. It represents an essential scientific solution for many of the environmental and societal challenges ahead of us, such as polymer degradation. Unlike traditional chemical methods, enzyme-mediated degradation is selective and environmentally friendly and requires milder conditions. Computational methods will play a critical role in developing such solutions by enabling more efficient bioprospecting of natural polymer-degrading enzymes.
pluriZyme
rational design
active site
protein engineering
computational chemistry
enzyme engineering
1. Introduction
Enzyme engineering is the design and construction of new enzymes or the modification of existing ones with desired properties for specific practical uses. It has widely proven its importance in a vast number of applications, with a significant recent increase in the number of studies in both academia and industry [1][2][3]. Such an increase is largely based on the advantages of using proteins to catalyze reactions, mainly in the form of sustainable and greener alternatives to chemical catalysts. In addition, the constantly growing supply of new enzymes derived from bioprospecting campaigns, along with their adaptable nature, makes them a very interesting raw material. In this sense, enzyme engineering aims to improve the performance of these natural catalysts in terms of properties such as activity, specificity, and stability.
2. How to Develop a PluriZyme
PluriZymes are proteins with more than one active site capable of enzymatic catalysis, where at least one of these has been designed by protein engineering. In this procedure, rational design plays a crucial role. The overall workflow for creating a de novo pluriZyme follows the steps summarized in Figure 1.
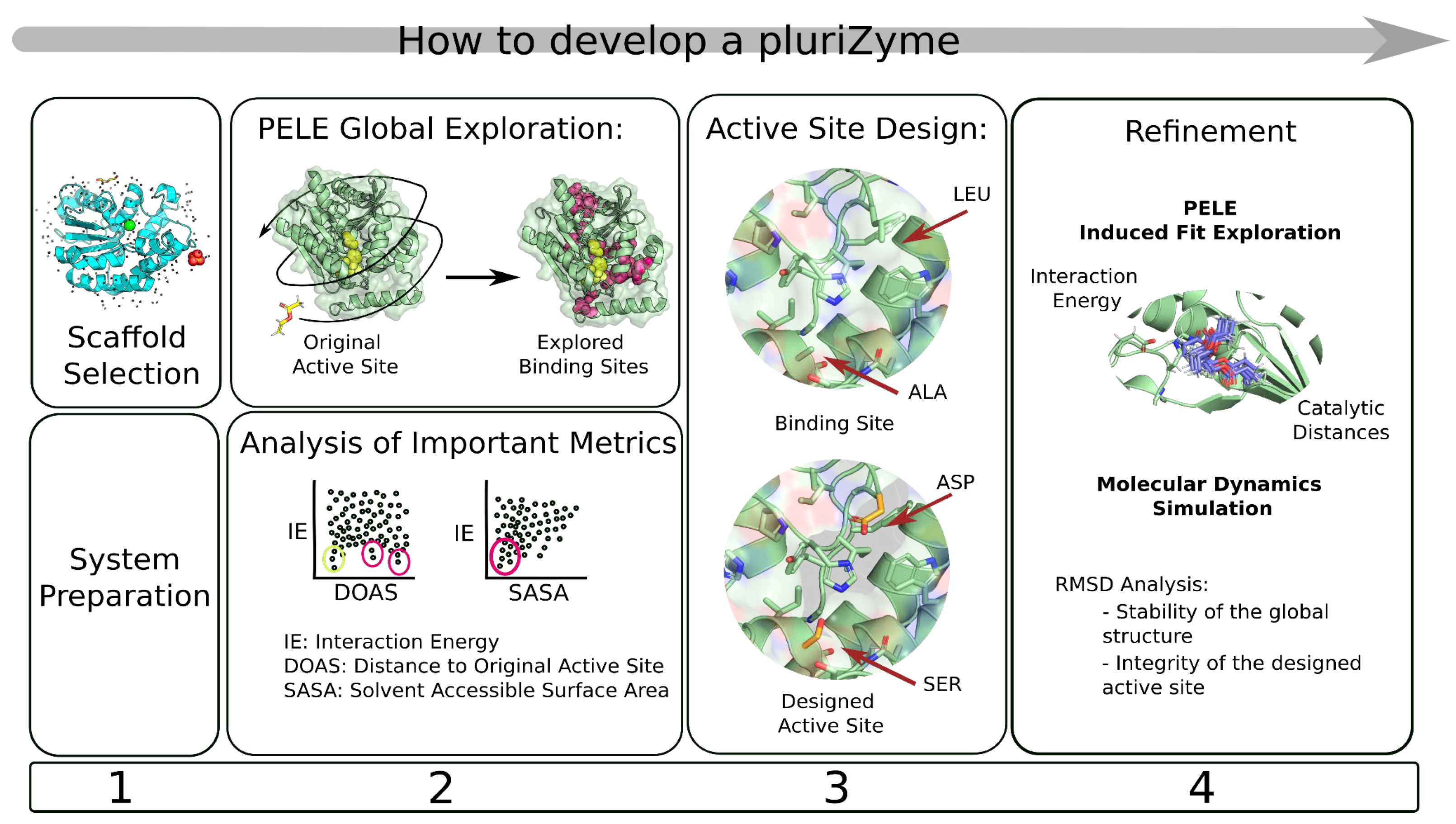
Figure 1. Workflow for the development of pluriZymes.
2.1. System Preparation
The first step always consists of knowing the system to prepare it adequately: the cellular location (membrane or soluble protein), number of system subunits, stability at different pHs and temperatures, cofactors, and modified or essential residues. The next step starts from a 3D protein structure that ideally comes from a resolved crystal or nuclear magnetic resonance (NMR). Researchers can also start from a model thanks to homology modeling and, more recently, to the breakthrough of deep learning structural builders, such as AlphaFold 2.0 [4]. Moreover, a model structure can add missing or omitted parts to the experimental structure. Finally, the hydrogen bond network should be optimized;researchers typically use Maestro’s Protein Preparation Wizard [5] to correct the protonation states depending on the pH of interest.
2.2. Binding Site Search
The second step, since researchers want to design new active sites in a protein scaffold, involves selecting a substrate(s) to simulate the active-site binding event. Once the substrate is selected, researchers prepare it with a quantum mechanics calculation in an implicit solvent and obtain the ESP charges with Jaguar [6]. Now that both the protein and the substrate are prepared, researchers perform PELE global exploration [7] (also known as SiteFinder), aimed at identifying potential binding sites (other than the wild-type active site if researchers are working with an enzyme) preexisting in the natural protein scaffold. PELE (Protein Energy Landscape Exploration) is the in-house Monte Carlo software capable of mapping complex intermolecular biophysical problems, such as global ligand migration, local induced fit, etc. [8]. As PELE uses implicit solvent (OBC or SGB), it tends to close the protein because the system maximizes stabilizing contacts between residues. Therefore, adding a series of constraints to the initial structure is necessary, generally with a force of 5 kcal/(mol·Å2) to one residue every 5 or 10 residues along the sequence. The metrics that most interest researchers at this point are the solvent-accessible surface area (SASA) of the substrate and the binding energy throughout the simulation. If researchers observe a local energy minimum, it might indicate that the substrate, during its exploration, has found a binding site—a cavity in which it can remain for a long time due to stabilizing intermolecular interactions. If researchers do not find a potential new binding site, researchers must consider whether researchers want to continue working with the system by designing a binding site—by opening/enlarging some nascent cavity, for example. For this, researchers might still want to identify some transient site by selecting the best interaction energies (nascent binding site) and enlarge it, similarly to what researchers performed recently to increase promiscuity in an esterase [9]. While researchers use PELE in the laboratory, other software capable of describing potential binding sites in an enzyme could be used at this point, such as a combination of docking and molecular dynamics [10][11] and even AI-based tools [12].
2.3. From Binding Site to Active Site
The third step involves the conversion of the binding site into an active site, involving amino acid mutagenesis to introduce a catalytic triad. When carrying out mutagenesis, researchers consider several factors, including: (i) the conservation of the residues that researchers want to mutate in other homologous sequences, (ii) prioritizing mutations to residues of the same category, (iii) prioritizing residues close in sequence space (not only in Euclidean space), and (iv) amino acid recycling. Regarding this last point, researchers prioritize catalytic triads where researchers use a wild-type acidic residue, since adding negative charges to a protein might have a detrimental effect. The presence of negative charges on acidic residues (such as aspartate or glutamate) can lead to unfavorable interactions with the carbonyl oxygens of the protein backbone. The extent of destabilization may vary depending on the distance to a neighboring backbone carbonyl. The introduction of a negative charge has the potential to impact stability, exerting both local and global effects [13].
Researchers' goal is to obtain an active site with well-organized catalytic triads (with proper distances and angles) and with the least possible number of mutations; for this, as many combinations as possible of potential catalytic triads are made.
Next, researchers perform a refinement simulation of the newly designed active site(s) using the likelihood of catalytic encounters using PELE simulations. researchers explore the movements of the substrate inside the cavity mapping and how mutations affect its binding energy profile and localization. Ideally, researchers want the substrate to remain in the binding site with similar or even better substrate-binding energies (the intermolecular interaction energy provided by PELE). Eventually, researchers analyze the distances between the reactive atoms of the substrate and the catalytic residues, accounting for all the catalytic events that can be observed. Researchers consider a catalytic event to occur when the catalytic distances between the substrate and the reactive residues are below ~4 Å. In an ester hydrolase, for example, it would correspond to the distance between the ester carbon and the alcoholic oxygen from the catalytic serine. Moreover, the rest of the catalytic residues must adopt reasonable distances and angles (including those that participate indirectly, such as the oxyanion holes in the case of hydrolases). The number of catalytic poses helps researchers to rank different catalytic constructs and assess the activity of the newly designed active site.
2.4. Refinement
Designs with better stability, binding energy, and number of catalytic events are further refined with molecular dynamics simulations to give more robustness to the engineering process. The goal is to double check the stability of the global structure, the integrity of the newly designed catalytic triad, and that the substrate does not abandon (escape) the active site. After the MD analysis, which is based on distance measurements and RMSD, a ranking of the mutants is established, and the best ones are selected for experimental validation.
2.5. Automation of PluriZyme Design
Out of the different steps involved in the pluriZyme design recipe (shown in Figure 1), the third step, active-site design, is the one that has involved the most human intervention and intuition in the different pluriZymes designed to date (see below). In order to alleviate the non-deterministic nature of the procedure, researchers developed automated software.
Toward this goal, a new Monte Carlo software program, AsiteDesign, was implemented and published recently [14]; it is mainly based on the pyRosetta library [15]. The algorithm starts by selecting a set of positions defined by the user, which should identify those residues around the identified alternative binding site. Then, all these positions are allowed to be mutated to create randomly different combinations of catalytic triads along the simulation, and the variants are ranked based on an energy metric after every iteration (Figure 2). Distance constraints are enforced during the simulation to provide the correct distances to the introduced catalytic residues. Ligand sampling is included in the simulation, as researchers believe it helps find the optimal solutions and ensures that the active site will have activity against that molecule. To enhance the sampling, adaptive reinforcement [8] and simulated annealing protocols are implemented. The number of designs increases by n!/(n − k)! (where k = 3 in catalytic triads). Thus, when the number of residues is 10 (n = 10), the number of possible catalytic triads is 720. This combinatorial problem is what AsiteDesign tries to address by intelligently sampling the possible combinations and outputting the best ones ranked by an energy metric.
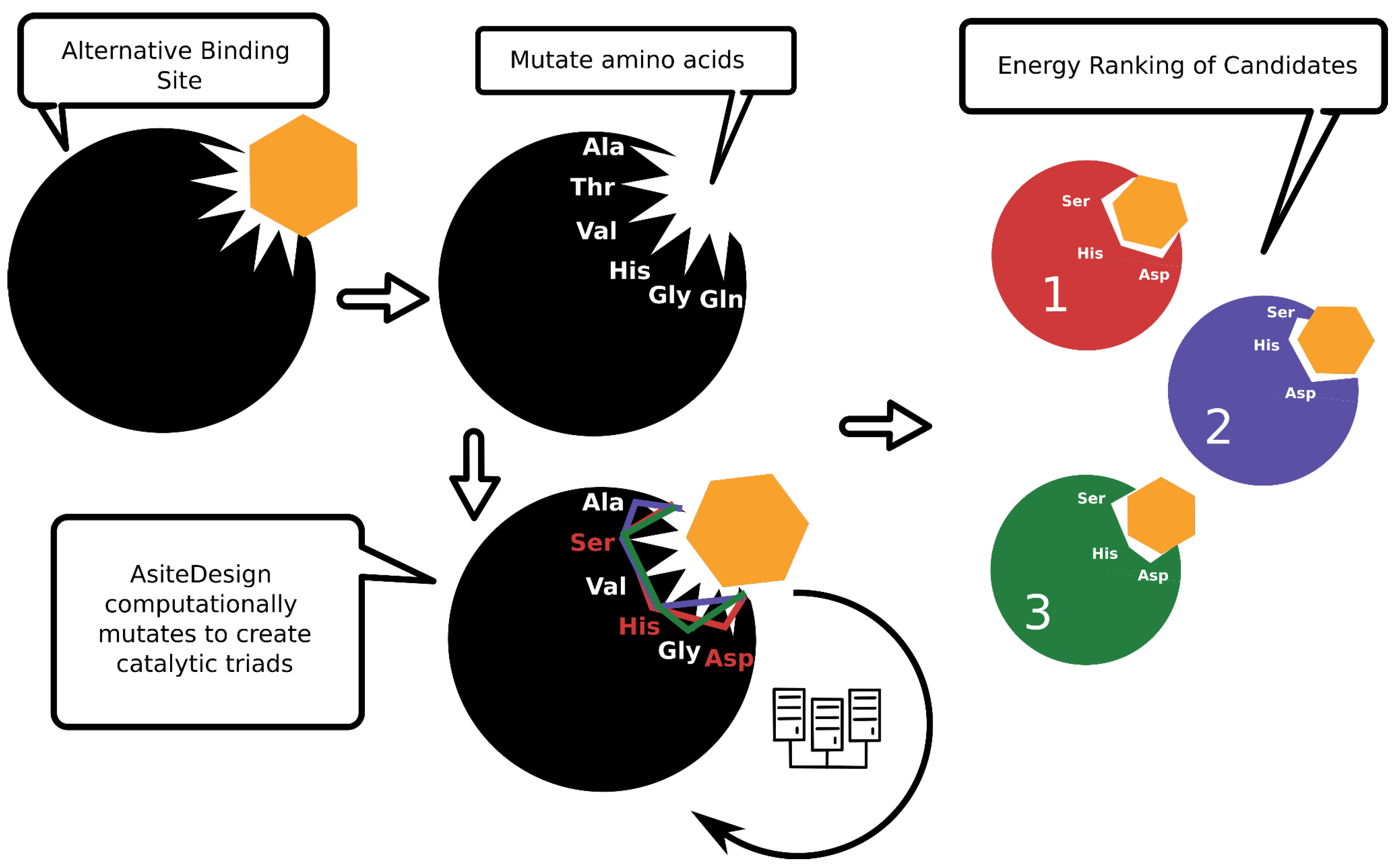
Figure 2. Simplified scheme of AsiteDesign workflow. The colors of the enzyme scaffold represent different mutants that AsiteDesign returns as possible options to insert a catalytic triad. The digits refer to the energy ranking of these designs.
AsiteDesign gives reliable catalytic triads without requiring the user to design them. In fact, the used benchmark was an esterase, and the catalytic residues of the system were mutated to Ala to see if the protocol could recover the original active site. AsiteDesign recovered the wild-type active site as the top-ranked solution. Likewise, it gave alternative catalytic triads of which the second-best option was assayed experimentally and had hydrolase activity [14]. Researchers have made the code freely available to everyone: a container version is available at https://github.com/BSC-CNS-EAPM/AsiteDesign-container, accessed on 21 December 2023; its usage, however, requires a Rosetta license (which is free for non-commercial use).
References
- Lovelock, S.L.; Crawshaw, R.; Basler, S.; Levy, C.; Baker, D.; Hilvert, D.; Green, A.P. The road to fully programmable protein catalysis. Nature 2022, 606, 49–58.
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. Engl. 2021, 60, 88–119.
- Arnal, G.; Anglade, J.; Gavalda, S.; Tournier, V.; Chabot, N.; Bornscheuer, U.T.; Weber, G.; Marty, A. Assessment of Four Engineered PET Degrading Enzymes Considering Large-Scale Industrial Applications. ACS Catal. 2023, 13, 13156–13166.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234.
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142.
- Acebes, S.; Fernandez-Fueyo, E.; Monza, E.; Lucas, M.F.; Almendral, D.; Ruiz-Dueñas, F.J.; Lund, H.; Martinez, A.T.; Guallar, V. Rational Enzyme Engineering Through Biophysical and Biochemical Modeling. ACS Catal. 2016, 6, 1624–1629.
- Lecina, D.; Gilabert, J.F.; Guallar, V. Adaptive simulations, towards interactive protein-ligand modeling. Sci. Rep. 2017, 7, 8466.
- Roda, S.; Fernandez-Lopez, L.; Cañadas, R.; Santiago, G.; Ferrer, M.; Guallar, V. Computationally Driven Rational Design of Substrate Promiscuity on Serine Ester Hydrolases. ACS Catal. 2021, 11, 3590–3601.
- Campbell, S.J.; Gold, N.D.; Jackson, R.M.; Westhead, D.R. Ligand binding: Functional site location, similarity and docking. Curr. Opin. Struct. Biol. 2003, 13, 389–395.
- Kua, J.; Zhang, Y.; McCammon, J.A. Studying Enzyme Binding Specificity in Acetylcholinesterase Using a Combined Molecular Dynamics and Multiple Docking Approach. J. Am. Chem. Soc. 2002, 124, 8260–8267.
- Rauer, C.; Sen, N.; Waman, V.P.; Abbasian, M.; Orengo, C.A. Computational approaches to predict protein functional families and functional sites. Curr. Opin. Struct. Biol. 2021, 70, 108–122.
- Pokkuluri, P.R.; Gu, M.; Cai, X.; Raffen, R.; Stevens, F.J.; Schiffer, M. Factors Contributing to Decreased Protein Stability When Aspartic Acid Residues Are in Beta-Sheet Regions. Protein Sci. 2002, 11, 1687–1694.
- Roda, S.; Terholsen, H.; Meyer, J.-R.-H.; Guallar, V.; Bornscheuer, U.; Kazemi, M. AsiteDesign: A semi-rational algorithm for automated enzyme design. J. Phys. Chem. B 2023, 127, 2661–2670.
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
734
Revisions:
3 times
(View History)
Update Date:
12 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No