 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jose-Angel Perez-Rivera | -- | 2318 | 2024-01-08 13:05:01 | | | |
| 2 | Peter Tang | Meta information modification | 2318 | 2024-01-09 06:14:54 | | |
Video Upload Options
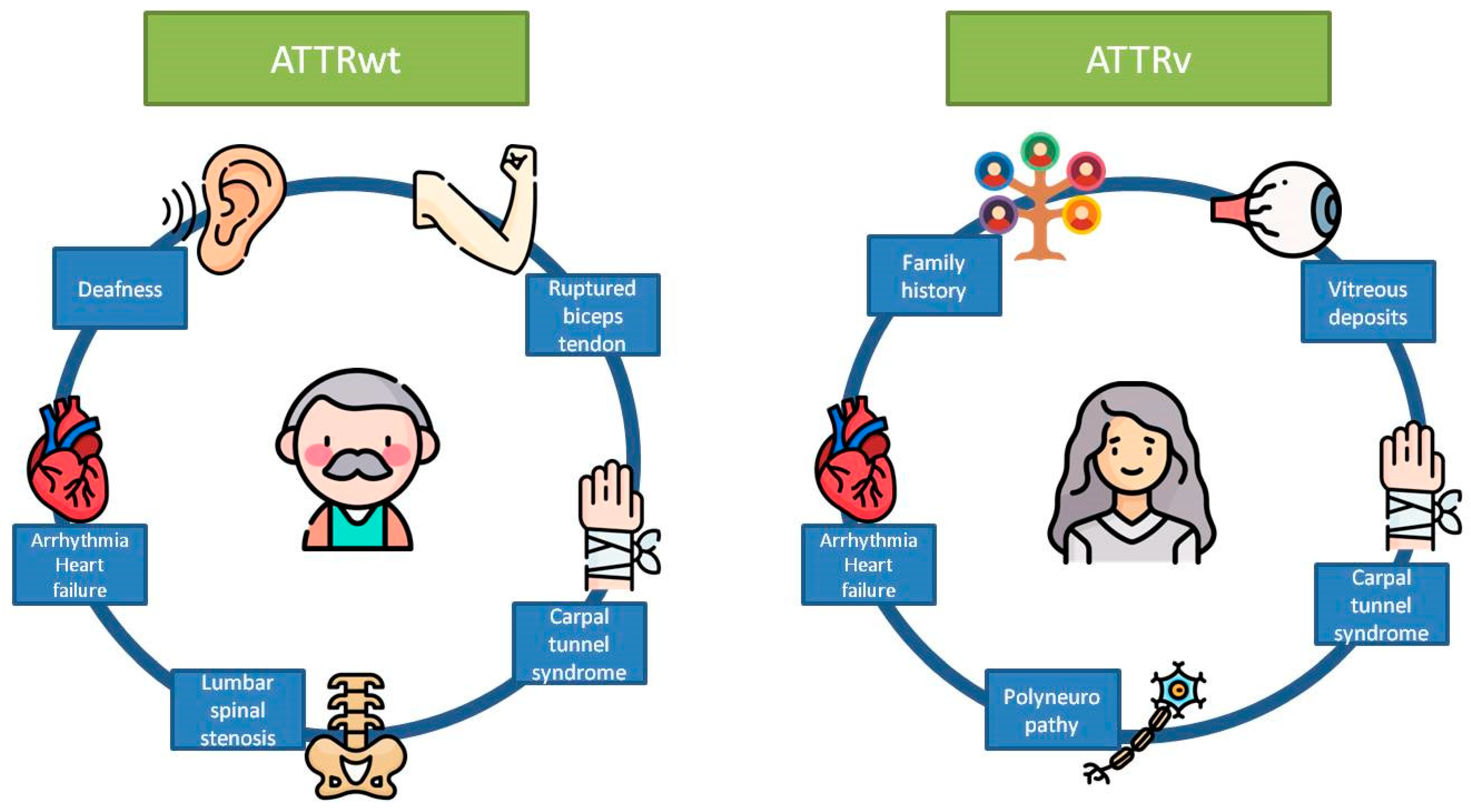
Transthyretin amyloid cardiomyopathy (ATTR-CM) is an increasingly diagnosed condition. Although wild-type transthyretin amyloidosis (ATTRwt) is the most common ATTR-CM, hereditary transthyretin amyloidosis (ATTRv) may also occur. Genetic testing for transthyretin pathogenic variants is recommended for patients with a confirmed clinical diagnosis of ATTR-CM. In fact, confirmation of this autosomal dominant pathogenic variant prompts genetic counselling and allows early identification of affected relatives. Additionally, in the presence of an ATTR-CM-associated polyneuropathy, specific drugs targeting transthyretin can be used.
1. Introduction
|
Localization |
Type |
Red Flag |
Amyloidosis Where It Is Most Frequently Found |
|---|---|---|---|
|
Extracardiac |
Clinical |
Ruptured biceps tendon |
ATTRwt |
|
Carpal tunnel syndrome |
ATTRwt and ATTRv |
||
|
Deafness |
ATTRwt |
||
|
Lumbar spinal stenosis |
ATTRwt |
||
|
Polyneuropathy |
ATTRv |
||
|
Vitreous deposits |
ATTRv |
||
|
Family history |
ATTRv |
||
|
Cardiac |
Clinical |
Heart failure |
ATTRwt and ATTRv |
|
Atrial fibrillation |
ATTRwt and ATTRv |
||
|
ECG |
Pseudoinfarct pattern |
ATTRwt and ATTRv |
|
|
Low QRS voltage |
ATTRwt and ATTRv |
||
|
Laboratory |
Disproportionally elevated NT-proBNP and troponin |
ATTRwt and ATTRv |
|
|
Echocardiogram |
Granular sparkling of myocardium |
ATTRwt and ATTRv |
|
|
Increased right ventricular wall thickness |
ATTRwt and ATTRv |
||
|
Increased valve thickness |
ATTRwt and ATTRv |
||
|
Pericardial effusion |
ATTRwt and ATTRv |
||
|
Reduced longitudinal strain with apical sparing pattern |
ATTRwt and ATTRv |
||
|
Cardiac magnetic resonance |
Abnormal gadolinium kinetics |
ATTRwt and ATTRv |
|
|
Elevated native T1 values |
ATTRwt and ATTRv |
||
|
Increased extracellular volume |
ATTRwt and ATTRv |

2. Utility of Family Screening
3. Utility of Genetic Testing in Clinical Characterization and Anticipation of Symptoms
4. Utility of Genetic Testing for the Initiation of a Specific Treatment
5. Utility of Genetic Testing in Prognosis Assessment
References
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and Treatment of Cardiac Amyloidosis. A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Heart Fail. 2021, 23, 512–526.
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of Transthyretin-Related Hereditary Amyloidosis for Clinicians. Orphanet J. Rare Dis. 2013, 8, 31.
- Ueda, M.; Ando, Y. Recent Advances in Transthyretin Amyloidosis Therapy. Transl. Neurodegener. 2014, 3, 19.
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-Type Transthyretin Amyloidosis as a Cause of Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2015, 36, 2585–2594.
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377.
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.-L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile Systemic Amyloidosis Affects 25% of the Very Aged and Associates with Genetic Variation in Alpha2-Macroglobulin and Tau: A Population-Based Autopsy Study. Ann. Med. 2008, 40, 232–239.
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020.
- González-López, E.; López-Sainz, Á.; Garcia-Pavia, P. Diagnosis and Treatment of Transthyretin Cardiac Amyloidosis. Progress and Hope. Rev. Esp. Cardiol. (Engl. Ed.) 2017, 70, 991–1004.
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.J.M.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M.; et al. Common Origin of the Val30Met Mutation Responsible for the Amyloidogenic Transthyretin Type of Familial Amyloidotic Polyneuropathy. J. Med. Genet. 2004, 41, e51.
- Zaros, C.; Genin, E.; Hellman, U.; Saporta, M.A.; Languille, L.; Wadington-Cruz, M.; Suhr, O.; Misrahi, M.; Planté-Bordeneuve, V. On the Origin of the Transthyretin Val30Met Familial Amyloid Polyneuropathy. Ann. Hum. Genet. 2008, 72, 478–484.
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T.; Kyriakides, T. European Network for TTR-FAP (ATTReuNET) Recommendations for Presymptomatic Genetic Testing and Management of Individuals at Risk for Hereditary Transthyretin Amyloidosis. Curr. Opin. Neurol. 2016, 29, S27–S35.
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary Transthyretin Amyloidosis Overview. Neurol. Sci. 2022, 43, 595–604.
- Sekijima, Y. Hereditary Transthyretin Amyloidosis. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993.
- Conceição, I.; Damy, T.; Romero, M.; Galán, L.; Attarian, S.; Luigetti, M.; Sadeh, M.; Sarafov, S.; Tournev, I.; Ueda, M. Early Diagnosis of ATTR Amyloidosis through Targeted Follow-up of Identified Carriers of TTR Gene Mutations. Amyloid 2019, 26, 3–9.
- Sekijima, Y. Transthyretin (ATTR) Amyloidosis: Clinical Spectrum, Molecular Pathogenesis and Disease-Modifying Treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043.
- Lemos, C.; Coelho, T.; Alves-Ferreira, M.; Martins-da-Silva, A.; Sequeiros, J.; Mendonça, D.; Sousa, A. Overcoming Artefact: Anticipation in 284 Portuguese Kindreds with Familial Amyloid Polyneuropathy (FAP) ATTRV30M. J. Neurol. Neurosurg. Psychiatry 2014, 85, 326–330.
- Adams, D.; Cauquil, C.; Theaudin, M.; Rousseau, A.; Algalarrondo, V.; Slama, M.S. Current and Future Treatment of Amyloid Neuropathies. Expert Rev. Neurother. 2014, 14, 1437–1451.
- Reinés, J.B.; Vera, T.R.; Martín, M.U.; Serra, H.A.; Campins, M.M.C.; Millán, J.M.D.; Lezaun, C.G.; Cruz, M.R. Epidemiology of Transthyretin-Associated Familial Amyloid Polyneuropathy in the Majorcan Area: Son Llàtzer Hospital Descriptive Study. Orphanet J. Rare Dis. 2014, 9, 29.
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016.
- Rapezzi, C.; Elliott, P.; Damy, T.; Nativi-Nicolau, J.; Berk, J.L.; Velazquez, E.J.; Boman, K.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; et al. Efficacy of Tafamidis in Patients with Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Analyses From ATTR-ACT. JACC Heart Fail. 2021, 9, 115–123.
- Mathew, V.; Wang, A.K. Inotersen: New Promise for the Treatment of Hereditary Transthyretin Amyloidosis. Drug Des. Dev. Ther. 2019, 13, 1515–1525.
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21.
- Di Stefano, V.; Fava, A.; Gentile, L.; Guaraldi, P.; Leonardi, L.; Poli, L.; Tagliapietra, M.; Vastola, M.; Fanara, S.; Ferrero, B.; et al. Italian Real-Life Experience of Patients with Hereditary Transthyretin Amyloidosis Treated with Patisiran. Pharmacogenomics Pers. Med. 2022, 15, 499–514.
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-Acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382.
- Griffin, J.M.; Rosenthal, J.L.; Grodin, J.L.; Maurer, M.S.; Grogan, M.; Cheng, R.K. ATTR Amyloidosis: Current and Emerging Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncology 2021, 3, 488–505.
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31.
- Benson, M.D.; Dasgupta, N.R.; Rissing, S.M.; Smith, J.; Feigenbaum, H. Safety and Efficacy of a TTR Specific Antisense Oligonucleotide in Patients with Transthyretin Amyloid Cardiomyopathy. Amyloid 2017, 24, 219–225.
- Monda, E.; Bakalakos, A.; Rubino, M.; Verrillo, F.; Diana, G.; De Michele, G.; Altobelli, I.; Lioncino, M.; Perna, A.; Falco, L.; et al. Targeted Therapies in Pediatric and Adult Patients With Hypertrophic Heart Disease: From Molecular Pathophysiology to Personalized Medicine. Circ. Heart Fail. 2023, 16, e010687.
- Russo, M.; Gentile, L.; Di Stefano, V.; Di Bella, G.; Minutoli, F.; Toscano, A.; Brighina, F.; Vita, G.; Mazzeo, A. Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area. Brain Sci. 2021, 11, 545.
- Falcão de Campos, C.; Conceição, I. Updated Evaluation of the Safety, Efficacy and Tolerability of Tafamidis in the Treatment of Hereditary Transthyretin Amyloid Polyneuropathy. Drug Healthc. Patient Saf. 2023, 15, 51–62.
- Gillmore, J.D.; Damy, T.; Fontana, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A New Staging System for Cardiac Transthyretin Amyloidosis. Eur. Heart J. 2018, 39, 2799–2806.
- Obi, C.A.; Mostertz, W.C.; Griffin, J.M.; Judge, D.P. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist Debakey Cardiovasc. J. 2022, 18, 17–26.

