 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ilya Akberdin | -- | 4146 | 2023-12-28 09:40:40 | | | |
| 2 | Mona Zou | + 1 word(s) | 4147 | 2023-12-29 05:32:11 | | | | |
| 3 | Mona Zou | -14 word(s) | 4133 | 2024-01-03 02:42:44 | | |
Video Upload Options
Methanotrophy is the ability of an organism to capture and utilize the greenhouse gas, methane, as a source of energy-rich carbon. Over the years, significant progress has been made in understanding of mechanisms for methane utilization, mostly in bacterial systems, including the key metabolic pathways, regulation and the impact of various factors (iron, copper, calcium, lanthanum, and tungsten) on cell growth and methane bioconversion. The implementation of -omics approaches provided vast amount of heterogeneous data that require the adaptation or development of computational tools for a system-wide interrogative analysis of methanotrophy. The genome-scale mathematical modeling of its metabolism has been envisioned as one of the most productive strategies for the integration of muti-scale data to better understand methane metabolism and enable its biotechnological implementation.
1. The Stages of Metabolic Model Reconstruction
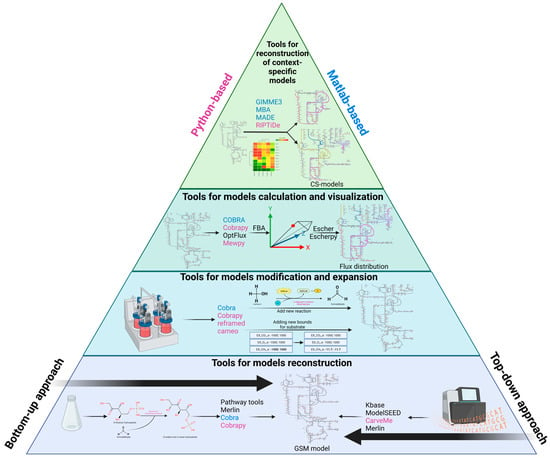
2. Databases of the Microorganisms’ Genomes
3. GSM Models for C1-Utilizing Bacteria
4. Web Resources and Tools for Automatic Reconstruction of GSM Models
4.1. Web Resources
4.2. GUI-Based Desktop Programs
4.3. Packages and Command Line Programs
2.5. Web-Resources and Tools for Analysis of GSM Models
References
- Feist, A.M.; Herrgård, M.J.; Thiele, I.; Reed, J.L.; Palsson, B. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009, 7, 129–143.
- Haggart, C.R.; Bartell, J.A.; Saucerman, J.J.; Papin, J.A. Whole-genome metabolic network reconstruction and constraint-based modeling. Methods Enzymol. 2011, 500, 411–433.
- Tang, Y.J.; Sapra, R.; Joyner, D.; Hazen, T.C.; Myers, S.; Reichmuth, D.; Blanch, H.; Keasling, J.D. Analysis of metabolic pathways and fluxes in a newly discovered thermophilic and ethanol-tolerant Geobacillus strain. Biotechnol. Bioeng. 2009, 102, 1377–1386.
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566–569.
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121.
- Edwards, J.S.; Covert, M.; Palsson, B. Metabolic modelling of microbes: The flux-balance approach. Environ. Microbiol. 2002, 4, 133–140.
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248.
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120.
- Cordova, L.T.; Long, C.P.; Venkataramanan, K.P.; Antoniewicz, M.R. Complete genome sequence, metabolic model construction and phenotypic characterization of Geobacillus LC300, an extremely thermophilic, fast growing, xylose-utilizing bacterium. Metab. Eng. 2015, 32, 74–81.
- Akberdin, I.R.; Thompson, M.; Hamilton, R.; Desai, N.; Alexander, D.; Henard, C.A.; Guarnieri, M.T.; Kalyuzhnaya, M.G. Methane utilization in Methylomicrobium alcaliphilum 20ZR: A systems approach. Sci. Rep. 2018, 8, 2512.
- Henard, C.A.; Akberdin, I.R.; Kalyuzhnaya, M.G.; Guarnieri, M.T. Muconic acid production from methane using rationally-engineered methanotrophic biocatalysts. Green Chem. 2019, 21, 6731–6737.
- Gupta, A.; Ahmad, A.; Chothwe, D.; Madhu, M.K.; Srivastava, S.; Sharma, V.K. Genome-scale metabolic reconstruction and metabolic versatility of an obligate methanotroph Methylococcus capsulatus str. Bath. PeerJ 2019, 7, e6685.
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2018, 20, 1085–1093.
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551.
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42.
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003.
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612.
- Vallenet, D.; Calteau, A.; Dubois, M.; Amours, P.; Bazin, A.; Beuvin, M.; Burlot, L.; Bussell, X.; Fouteau, S.; Gautreau, G.; et al. MicroScope: An integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 2020, 48, D579–D589.
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.J.; Webb, C.; Wu, D.; et al. The IMG/M data management and analysis system v.7: Content updates and new features. Nucleic Acids Res. 2023, 51, D723–D732.
- Karp, P.D.; Midford, P.E.; Billington, R.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Ong, W.K.; Subhraveti, P.; Caspi, R.; Fulcher, C.; et al. Pathway Tools version 23.0 update: Software for pathway/genome informatics and systems biology. Brief Bioinform. 2021, 22, 109–126.
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muñiz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077.
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280.
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462.
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531.
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508.
- Wittig, U.; Rey, M.; Weidemann, A.; Kania, R.; Müller, W. SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 2018, 46, D656–D660.
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Sherry, S.T.; Yankie, L.; Karsch-Mizrachi, I. GenBank 2023 update. Nucleic Acids Res. 2023, 51, D141–D144.
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808.
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591.
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, annotation, and comparative genomics in PATRIC, the all bacterial bioinformatics resource center. Methods Mol. Biol. 2018, 1704, 79–101.
- Akberdin, I.R.; Thompson, M.; Kalyuzhnaya, M.G. Systems biology and metabolic modeling of C1-metabolism. In Methane Biocatalysis: Paving the Way to Sustainability; Springer: Cham, Switzerland, 2018.
- Kabimoldayev, I.; Nguyen, A.D.; Yang, L.; Park, S.; Lee, E.Y.; Kim, D. Basics of genome-scale metabolic modeling and applications on C1-utilization. FEMS Microbiol. Lett. 2018, 365, fny241.
- Nguyen, A.D.; Lee, E.Y. Engineered Methanotrophy: A Sustainable Solution for Methane-Based Industrial Biomanufacturing. Trends Biotechnol. 2021, 39, 381–396.
- Guo, S.; Nguyen, D.T.N.; Chau, T.H.T.; Fei, Q.; Lee, E.Y. Systems Metabolic Engineering of Methanotrophic Bacteria for Biological Conversion of Methane to Value-Added Compounds. Adv. Biochem. Eng./Biotechnol. 2022, 180, 91–126.
- Torre, A.; Metivier, A.; Chu, F.; Laurens, L.M.L.; Beck, D.A.C.; Pienkos, P.T.; Lidstrom, M.E.; Kalyuzhnaya, M.G. Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb. Cell Fact. 2015, 14, 188.
- Demidenko, A.; Akberdin, I.R.; Allemann, M.; Allen, E.E.; Kalyuzhnaya, M.G. Fatty acid biosynthesis pathways in Methylomicrobium buryatense 5G(B1). Front. Microbiol. 2017, 7, 2167.
- Kalyuzhnaya, M.G.; Yang, S.; Rozova, O.N.; Smalley, N.E.; Clubb, J.; Lamb, A.; Gowda, G.A.N.; Raftery, D.; Fu, Y.; Bringel, F.; et al. Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat. Commun. 2013, 4, 2785.
- Akberdin, I.R.; Collins, D.A.; Hamilton, R.; Oshchepkov, D.Y.; Shukla, A.K.; Nicora, C.D.; Nakayasu, E.S.; Adkins, J.N.; Kalyuzhnaya, M.G. Rare Earth Elements alter redox balance in Methylomicrobium alcaliphilum 20ZR. Front. Microbiol. 2018, 9, 2735.
- Lieven, C.; Petersen, L.A.H.; Jørgensen, S.B.; Gernaey, K.V.; Herrgard, M.J.; Sonnenschein, N. A Genome-Scale Metabolic Model for Methylococcus capsulatus (Bath) Suggests Reduced Efficiency Electron Transfer to the Particulate Methane Monooxygenase. Front. Microbiol. 2018, 9, 2947.
- Bordel, S.; Rodríguez, Y.; Hakobyan, A.; Rodríguez, E.; Lebrero, R.; Muñoz, R. Genome scale metabolic modeling reveals the metabolic potential of three Type II methanotrophs of the genus Methylocystis. Metab. Eng. 2019, 54, 191–199.
- Bordel, S.; Rojas, A.; Muñoz, R. Reconstruction of a Genome Scale Metabolic Model of the polyhydroxybutyrate producing methanotroph Methylocystis parvus OBBP. Microb. Cell Fact. 2019, 18, 104.
- Bordel, S.; Crombie, A.T.; Muñoz, R.; Murrell, J.C. Genome Scale Metabolic Model of the versatile methanotroph Methylocella silvestris. Microb. Cell Fact. 2020, 19, 144.
- Villada, J.C.; Duran, M.F.; Lim, C.K.; Stein, L.Y.; Lee, P.K.H. Integrative Genome-Scale Metabolic Modeling Reveals Versatile Metabolic Strategies for Methane Utilization in Methylomicrobium album BG8. mSystems 2022, 7, e0007322.
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158.
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A Web Application for Building, Sharing, and Embedding Data-Rich Visualizations of Biological Pathways. PLoS Comput. Biol. 2015, 11, e1004321.
- Devoid, S.; Overbeek, R.; DeJongh, M.; Vonstein, V.; Best, A.A.; Henry, C. Automated genome annotation and metabolic model reconstruction in the SEED and model SEED. Methods Mol. Biol. 2013, 985, 17–45.
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588.
- Faria, J.P.; Liu, F.; Edirisinghe, J.N.; Gupta, N.; Seaver, S.M.D.; Freiburger, A.P.; Zhang, Q.; Weisenhorn, P.; Conrad, N.; Zarecki, R.; et al. ModelSEED v2: High-throughput genome-scale metabolic model reconstruction with enhanced energy biosynthesis pathway prediction. bioRxiv 2023.
- Boele, J.; Olivier, B.G.; Teusink, B. FAME, the Flux Analysis and Modeling Environment. BMC Syst. Biol. 2012, 6, 8.
- Feng, X.; Xu, Y.; Chen, Y.; Tang, Y.J. MicrobesFlux: A web platform for drafting metabolic models from the KEGG database. BMC Syst. Biol. 2012, 6, 94.
- Capela, J.; Lagoa, D.; Rodrigues, R.; Cunha, E.; Cruz, F.; Barbosa, A.; Bastos, J.; Lima, D.; Ferreira, E.C.; Rocha, M.; et al. merlin, an improved framework for the reconstruction of high-quality genome-scale metabolic models. Nucleic Acids Res. 2022, 50, 6052–6066.
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 2020, 48, D402–D406.
- Olivier, B.G.; Mendoza, S.; Molenaar, D.; Teusink, B. MetaDraft Release: 0.9.5 2020. Available online: https://zenodo.org/records/4291058 (accessed on 12 December 2023).
- Liao, Y.C.; Tsai, M.H.; Chen, F.C.; Hsiung, C.A. GEMSiRV: A software platform for GEnome-scale metabolic model simulation, reconstruction and visualization. Bioinformatics 2012, 28, 1752–1758.
- Poolman, M.G. ScrumPy: Metabolic modelling with Python. IEE Proc. Syst. Biol. 2006, 153, 375–378.
- Aite, M.; Chevallier, M.; Frioux, C.; Trottier, C.; Got, J.; Cortés, M.P.; Mendoza, S.N.; Carrier, G.; Dameron, O.; Guillaudeux, N.; et al. Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLoS Comput. Biol. 2018, 14, e1006146.
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 2021, 22, 81.
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018, 19, 467.
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541.
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553.
- Saadat, N.P.; van Aalst, M.; Ebenhöh, O. Network Reconstruction and Modelling Made Reproducible with moped. Metabolites 2022, 12, 275.
- Jenior, M.L.; Glass, E.M.; Papin, J.A. Reconstructor: A COBRApy compatible tool for automated genome-scale metabolic network reconstruction with parsimonious flux-based gap-filling. Bioinformatics 2023, 39, btad367.
- Vezina, B.; Watts, S.C.; Hawkey, J.; Cooper, H.B.; Judd, L.M.; Jenney, A.; Monk, J.M.; Holt, K.E.; Wyres, K.L. Bactabolize: A tool for high-throughput generation of bacterial strain-specific metabolic models. bioRxiv 2023.
- Belcour, A.; Got, J.; Aite, M.; Delage, L.; Collén, J.; Frioux, C.; Leblanc, C.; Dittami, S.M.; Blanquart, S.; Markov, G.V.; et al. Inferring and comparing metabolism across heterogeneous sets of annotated genomes using AuCoMe. Genome Res. 2023, 33, 972–987.
- Hucka, M.; Bergmann, F.T.; Dräger, A.; Hoops, S.; Keating, S.M.; Le Novère, N.; Myers, C.J.; Olivier, B.G.; Sahle, S.; Schaff, J.C.; et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core. J. Integr. Bioinform. 2018, 15, 20170081.
- Prigent, S.; Frioux, C.; Dittami, S.M.; Thiele, S.; Larhlimi, A.; Collet, G.; Gutknecht, F.; Got, J.; Eveillard, D.; Bourdon, J.; et al. Meneco, a Topology-Based Gap-Filling Tool Applicable to Degraded Genome-Wide Metabolic Networks. PLoS Comput. Biol. 2017, 13, e1005276.
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702.
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74.
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45.
- Vilaça, P.; Maia, P.; Giesteira, H.; Rocha, I.; Rocha, M. Analyzing and designing cell factories with OptFlux. Methods Mol. Biol. 2018, 1716, 37–76.
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935.
- Shen, F.; Sun, R.; Yao, J.; Li, J.; Liu, Q.; Price, N.D.; Liu, C.; Wang, Z. Optram: In-silico strain design via integrative regulatory-metabolic network modeling. PLoS Comput. Biol. 2019, 15, e1006835.
- Pereira, V.; Cruz, F.; Rocha, M. MEWpy: A computational strain optimization workbench in Python. Bioinformatics 2021, 37, 2494–2496.
- Kim, M.K.; Lane, A.; Kelley, J.J.; Lun, D.S. E-Flux2 and sPOT: Validated methods for inferring intracellular metabolic flux distributions from transcriptomic data. PLoS ONE 2016, 11, e0157101.
- Kelley, J.J.; Maor, S.; Kim, M.K.; Lane, A.; Lun, D.S. MOST-Visualization: Software for producing automated textbook-style maps of genome-scale metabolic networks. Bioinformatics 2017, 33, 2596–2597.
- Mao, Z.; Yuan, Q.; Li, H.; Hang, Y.; Huang, Y.; Yang, C.; Wang, R.; Yang, Y.; Wu, Y.; Yang, S.; et al. CAVE: A cloud-based platform for analysis and visualization of metabolic pathways. Nucleic Acids Res. 2023, 51, W70–W77.
- Hari, A.; Lobo, D. Fluxer: A web application to compute, analyze and visualize genome-scale metabolic flux networks. Nucleic Acids Res. 2020, 48, W427–W435.
- Cardoso, J.G.R.; Jensen, K.; Lieven, C.; Hansen, A.S.L.; Galkina, S.; Beber, M.; Özdemir, E.; Herrgård, M.J.; Redestig, H.; Sonnenschein, N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018, 7, 1163–1166.
- Machado, D.; Ochsner, N.; Colpo, R.A. cdanielmachado/reframed: 1.4.0 2023. Available online: https://zenodo.org/records/7778365 (accessed on 12 December 2023).


