Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Piero Leone | -- | 2234 | 2023-12-18 12:01:54 | | | |
| 2 | Rita Xu | Meta information modification | 2234 | 2023-12-19 03:06:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tummolo, A.; Carella, R.; De Giovanni, D.; Paterno, G.; Simonetti, S.; Tolomeo, M.; Leone, P.; Barile, M. Inherited Metabolic Disorders Requiring Diet Regimen. Encyclopedia. Available online: https://encyclopedia.pub/entry/52874 (accessed on 27 July 2026).
Tummolo A, Carella R, De Giovanni D, Paterno G, Simonetti S, Tolomeo M, et al. Inherited Metabolic Disorders Requiring Diet Regimen. Encyclopedia. Available at: https://encyclopedia.pub/entry/52874. Accessed July 27, 2026.
Tummolo, Albina, Rosa Carella, Donatella De Giovanni, Giulia Paterno, Simonetta Simonetti, Maria Tolomeo, Piero Leone, Maria Barile. "Inherited Metabolic Disorders Requiring Diet Regimen" Encyclopedia, https://encyclopedia.pub/entry/52874 (accessed July 27, 2026).
Tummolo, A., Carella, R., De Giovanni, D., Paterno, G., Simonetti, S., Tolomeo, M., Leone, P., & Barile, M. (2023, December 18). Inherited Metabolic Disorders Requiring Diet Regimen. In Encyclopedia. https://encyclopedia.pub/entry/52874
Tummolo, Albina, et al. "Inherited Metabolic Disorders Requiring Diet Regimen." Encyclopedia. Web. 18 December, 2023.
Copy Citation
Many inherited metabolic disorders (IMDs), including disorders of amino acid, fatty acid, and carbohydrate metabolism, are treated with a dietary reduction or exclusion of certain macronutrients, putting one at risk of a reduced intake of micronutrients.
micronutrients
oligoelements
vitamins
inherited metabolic disorders
1. Introduction
Trace elements and vitamins, called together “micronutrients”, are essential components of human nutrition in health and disease [1][2]. They play a variety of biochemical roles as cofactors and coenzymes in metabolism, as antioxidants, and in genetic regulation and protein folding, being crucial for maintaining tissue function and metabolism [3].
For the general population, international recommendations for micronutrient intake are available in the form of Recommended Dietary Allowances (RDA) or Dietary Reference Intakes (DRI), but the impact of micronutrient deficiencies in disease settings remains limited [4].
The roles of water-soluble vitamins in cellular metabolism have been clarified for many years. In fact, vitamin-derived cofactors intervene in a series of biochemical reactions and, consistently, their genetically determined deficiencies, at various levels, are linked to clinical pictures of variable severity, sometimes with serious and fatal results [5][6][7][8][9][10].
It should also be mentioned that inherited metabolic disorders (IMDs), when treated with a specific diet, may result in secondary vitamin deficiencies. The major therapy strategy for a significant number of IMDs essentially involves a specialized diet treatment, requiring the reduction or exclusion of certain macronutrients, focused on the enzyme deficiency causing the disorder [11][12].
On the one hand, this regimen makes it possible to reduce the effects of the enzymatic defect, significantly improving the clinical outcome of the disorder; on the other hand, a selective diet, especially at the growing age, could be associated with a reduced intake of micronutrients. To escape this problem, over the years, guidelines for the management of different IMDs have reported recommendations on the integration of vitamins and trace elements, aimed at reducing this nutritional risk [13][14][15][16].
2. IMDs Requiring Special Diets
Nutritional therapy in IMDs is based on the basic principle of reducing the concentrations of toxic substrates by reducing the assumption of nutrients that produce them or by increasing their excretion while providing deficient products through supplementation. This approach is necessary for the normal growth and development of patients affected by several IMDs [17]. Special medical foods that include macro- and micronutrients but omit the offending substrate are available to help prevent such deficiencies. In addition to medical foods, other specialized nutritional products, including high doses of vitamins and amino acids, may be used in the management of IMDs.
Table 1 reports the main IMDs requiring a special diet, distinguishing them by type of disorder and the principal category of limited food.
Table 1. IMDs requiring special diet and associated micronutrient deficiency.
| Category of Disorder | Type of Disorder | Diet Regimen | Principal Category of Limited Food | Ref. |
|---|---|---|---|---|
| Amino acid disorders | Phenylketonuria | Low Phenylalanine intake | Meat, fish, eggs, pulses, milk and dairy products, cereals | [13] |
| Organic acidosis (OA) | Low natural protein intake | Meat, fish, eggs, pulses, milk and dairy products, cereals |
[14][18] | |
| UCDs | Low natural protein intake | Meat, fish, eggs, pulses, milk and dairy products, cereals |
[15] | |
| Fatty acid oxidation disorders | VLCADD | Low intake of long-chain fatty acids | Full-fat and semi-skimmed milk, egg yolks, fatty fish and meat, cheese, butter, margarine, vegetable oil, dried fruit, oilseeds, chocolate, baked products, industrial products | [16] |
| Carbohydrate disorders | Galactosemia | Galactose-restricted diet | Milk and derivatives | [19] |
| Hereditary fructose intolerance | Minimal fructose and absolute exclusion of sucrose and alimentary additives like caramel (E150), sweeteners isomalt (E963), maltitol (E965) mannitol (E421), sorbitol (E420), xylitol (E967) intake | Fruit, honey, vegetables, other products containing sugar | [20] | |
| Glycogen storage disorders (I, III, VI, IX) | Fructose, sucrose, and galactose exclusion (I) Moderately high protein and low sugar intake (III) Low carbohydrate intake (VII) |
Fruit, honey, vegetables, products containing sugar | [21][22] | |
| IMD treated with ketogenic diet | GLUT1 deficiency PDH deficiency |
Low carbohydrates and high fat intake |
Fruit, dessert pastry, sweets, juice, pasta, cereals and baked products, potatoes, pulses | [23][24] |
2.1. Disorders of Amino acid Metabolism
2.1.1. Phenylketonuria (PKU)
Phenylketonuria (PKU) is a rare inherited metabolic disorder characterized by the partial or total inability to convert the essential amino acid Phenylalanine (Phe) into Tyrosine (Tyr) due to biallelic pathogenetic mutations of the liver enzyme phenylalanine hydroxylase (PAH). If PKU is detected at birth and treated with a Phe-restricted diet, the neurological sequaele secondary to Phe accumulation can be controlled [13].
The Phe-restricted diet requires strict monitoring of patients’ nutritional status according to the PKU severity and type of diet [25][26]. The majority of patients, with the exception of those with mild hyperphenylalaninemia, consume little animal protein and mostly low natural protein diets. Therefore, supplemented Phe-free L-amino acids or formulations with no or little Phe content, such as Glycomacropeptides (GMP), are the main sources of micronutrients [27]. The necessary daily intake of micronutrients can be obtained by regularly assuming these formulations [13] (Figure 1).
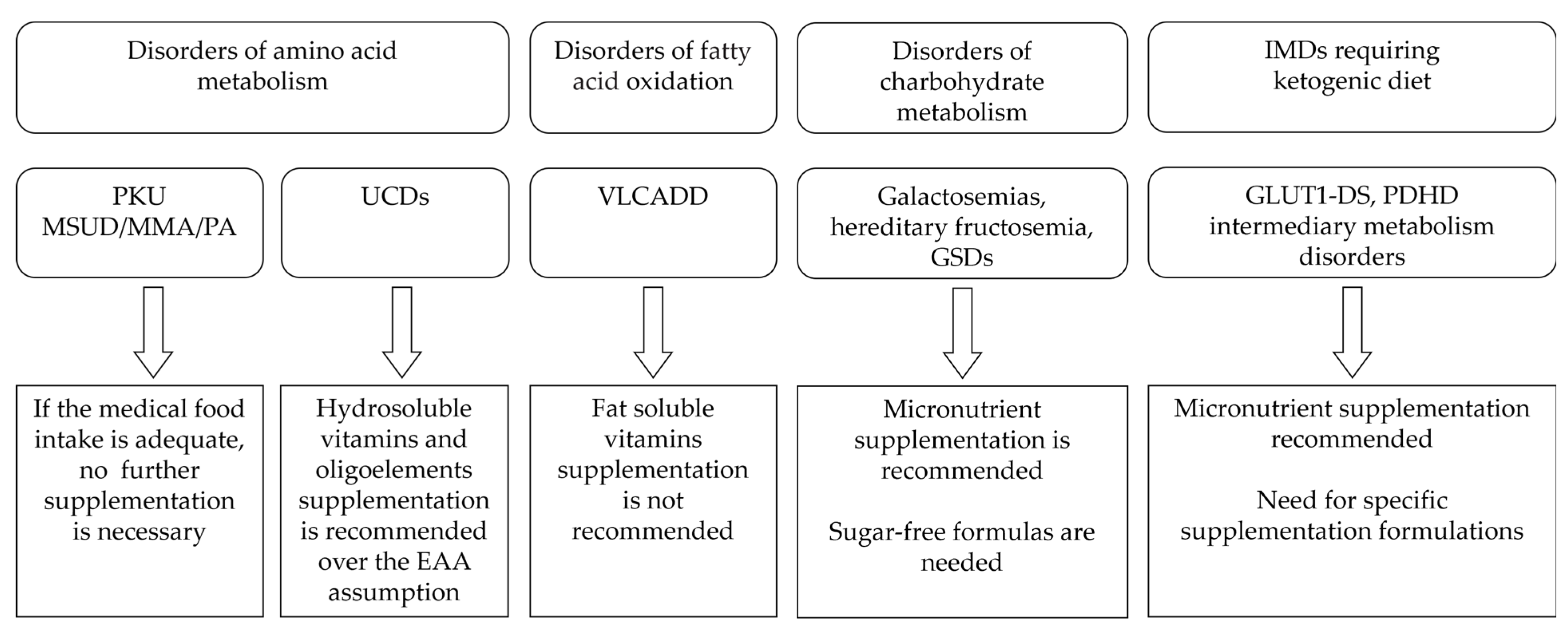
Figure 1. Main recommendations regarding micronutrient supplementation in different IMDs.
However, clinical symptoms of micronutrient deficiency are rarely reported, being mainly described for vitamin B12 deficiency, particularly after reducing or stopping micronutrient supplements or Phe-free L-amino acid supplements while following a vegan-like diet [28][29].
Studies by Evans et al. [30] and de Almeida et al. [31] showed that more than 90% of treated patients had adequate and normal ferritin levels. Crujeras et al. reported lower-than-normal selenium levels in 95% of PKU patients [32].
A study on the nutritional characteristics of adult PKU patients, according to their dietary adherence, reported that all patients in the adherent group met the Lower Reference Nutrient Intakes for the vast majority of micronutrients assessed. Nonadherent patients had significantly lower intakes of thiamine, riboflavin, niacin, vitamin B6, and phosphorus [33].
The literature review revealed poor data related to riboflavin and pyridoxine status in subjects undergoing a protein-restricted diet. Some old case series, by measuring the plasma pyridoxal 5′-phosphate (PLP), report on differences in pyridoxine metabolism in PKU children compared to healthy subjects, raising the need for personalized supplementation in this group of patients [34]. Children with PKU also showed an increase in the FAD effect and a concurrent decrease in glutathione reductase activity upon stopping group B vitamin therapy [35]. These findings are indicative of an inadequate riboflavin status. Since functional and direct biomarkers can be used in clinical practice to evaluate the levels of these two vitamins [36][37], it is necessary to reevaluate the patients’ pyridoxine and riboflavin status.
2.1.2. Maple Syrup Urine Disease, Propionic and Methylmalonic Acidemia
Maple syrup urine disease (MSUD), methylmalonic acidemia (MMA), and propionic acidemia (PA) are rare, autosomal recessive, multisystemic inborn errors of branched-chain amino acid metabolism, treated with a low-protein diet, precursor-free amino acid and/or isoleucine/valine supplementation [38].
The most recent guidelines for the above disorders emphasize the need for regular monitoring of micronutrient statuses to ensure adequate micronutrient intake [18]. As a matter of fact, most amino acid-free medical foods are supplemented with nutrients and micronutrients that may be deficient in a low-protein or low-precursor amino acid diet regimen. These formulas are usually supplemented with essential fatty acids, docosahexaenoic acid (DHA), vitamin D, vitamin A, calcium, iron, zinc, and selenium. Compliance with a full medical food prescription is important to meet these nutrient requirements [39].
Nutritional deficiencies have also been described for selenium and thiamine [40], secondary to the low animal protein intake. In addition, high-dose vitamin E and Coenzyme Q10 [18] are administered in order to prevent or treat optic neuropathy, which may alter visual acuity in MMA and PA patients [41][42].
2.1.3. Urea Cycle Disorders
Urea cycle disorders (UCDs) are a group of IMDs caused by a loss of function in one of the enzymes responsible for ureagenesis [43]. Long-term management of UCDs aims to prevent hyperammonemia and ensure normal development by the use of vitamin and mineral supplements, low-protein diets, essential amino acid supplements, and ammonia scavengers [15].
Supplementation is necessary for UCD patients on low-protein diets because of the risk of vitamin and mineral deficiencies, particularly iron, zinc, copper, calcium, and cobalamin [44][45].
In early-diagnosed patients, vitamin and mineral supplementations are generally started at weaning, in concomitance with milk intake reduction. Late-onset patients who are on a self-selected low-protein diet usually need vitamin and mineral supplements and regular dietary assessments [15].
2.2. Disorders of Fatty Acid Oxidation
Fatty acid oxidation disorders (FAOD) are a group of IMDs characterized by the defective transport or β-oxidation of fatty acids and are particularly involved in producing energy during fasting and stress episodes [46][47].
Patients affected by very-long-chain Acyl CoA dehydrogenase deficiency (VLCADD), one of the most severe forms of FAOD, undergo a dietary long-chain fatty acid restriction. Since they are susceptible to deficits in essential fatty acids and fat-soluble micronutrients [48], they should be evaluated for both. These patients may require supplementation with DHA or oils rich in essential fatty acids, such as linoleic acid and α-linoleic acid, to meet their nutritional needs. However, there are no reports regarding vitamin supplementation in subjects with long-chain fatty acid restriction. Although lower than normal levels of fat-soluble vitamins have been reported, recommendations for their supplementation cannot be made at this time [49].
2.3. Disorders of Carbohydrate Metabolism
2.3.1. Galactosemias
Galactosemias are a group of four hereditary disorders of galactose metabolism [50]. The most common form is Galactosemia type 1 due to deficiency of Galactose 1-phosphate urydyltransferase (GALT), which catalyzes one of the four reactions in the Leloir pathway, which converts galactose into glucose [51]. Diet is the cornerstone of the treatment of galactosemias, aimed at minimizing galactose intake [52][53].
An annual dietary assessment of calcium and vitamin D intake with measurement of plasma total 25-OH-vitamin D levels is recommended. Both calcium and vitamin D should be supplemented as necessary, following the age-specific recommendations for the general population.
Supplementation with vitamin K might be beneficial when combined with an adequate intake of calcium and vitamin D, but currently there is not enough evidence to recommend the routine use of vitamin K [19].
2.3.2. Hereditary Fructosemia
Dietary restriction of fructose, sucrose, sucralose, and sorbitol is the cornerstone of treatment for hereditary fructosemia (HF), an IMD caused by a deficiency in aldolase B (fructose-1,6-bisphosphate aldolase), which is responsible for the cleavage of fructose-1-phosphate [54]. Since fruit and vegetable intake is a dietary requirement, micronutrient deficiencies, particularly of water-soluble vitamins, are likely. However, there is great heterogeneity in vitamin supplementation practices among specialized centers.
In a recent report [20], most of the HF participants presented vitamin C (96.7%) and folate (90%) dietary intake below the recommended population reference. Up to 69% of the participants received vitamin C supplementation and 50% received folic acid supplementation. The amount of vitamin C supplementation correlated positively with correspondent plasma levels. Furthermore, non-supplemented HF patients were vitamin C deficient, with a statistically significant difference with respect to supplemented HF patients and healthy controls. Ensuring adequate vitamin supplementation in a disease requiring a reduction in fruit and vegetable intake is imperative [55]; supplementation with “sugar-free” multivitamin formulations is recommended.
2.3.3. Glycogen Storage Disorders (GSDs)
Liver glycogenosis: GSDI and III, GSDVI, and liver GSDIXs are a group of rare conditions due to a genetic enzymatic defect in the metabolism of glycogen [56]. They have in common hepatomegaly and hypoglycemia and undergo an overlapping dietetic approach. Although there is no consensus regarding the restriction of sugars in the diet, sucrose (fructose and glucose) and lactose (galactose and glucose) are often limited or avoided [21]. The most common among GSDs is GSDI, in which, as a result of the deficiency of glucose-6-phosphatase, fructose and galactose are not metabolized to glucose-6-phosphate [57][58].
Restricting fruit, juice, and dairy foods impacts two entire food groups and renders the diet inadequate. Careful assessment and supplementation of micronutrients are therefore required to avoid nutrient deficiencies. In a recent study, 61.5% of patients with GSDI who were tested for 25-OH-vitamin D levels were found to have insufficient levels (<30 ng/mL), despite their reported good compliance with prescribed supplements [22].
The restricted nature of the diet, aimed at maintaining normoglycemia, may also result in poor intake of iron, vitamin B12, and folic acid. In liver GSDs and in particular in GSDI, a complete multivitamin with mineral supplementation is essential. Without appropriate supplements, these patients are at risk of a variety of nutritional deficiencies.
2.4. IMD Requiring Ketogenic Diet
A ketogenic diet (KD) is characterized by a diet with a low carbohydrate, high fat, and a defined or variable protein content [23]. There are two main types of KD: the classical diet, which uses long-chain triglycerides as its primary fat source, and the medium-chain triglyceride (MCT) diet, which allows more carbohydrate and protein because of the increased ketogenic potential of MCT [59].
KD represents the recommended treatment for pyruvate dehydrogenase complex (PDHc) deficiency and glucose transporter type 1 deficiency syndrome (GLUT1-DS) as it directly targets the underlying metabolic condition.
In other IMDs, mainly of intermediary metabolism, such as glycogen storage diseases and disorders of mitochondrial energy supply, supplementation with ketone bodies may ameliorate clinical symptoms and laboratory parameters [37][60][61].
Side effects have been classically reported, including specific micronutrient deficiencies in vitamin D and calcium, vitamin C, thiamine, and selenium [62][63][64]. The KD should be supplemented with vitamins, minerals, and trace elements, with plasma levels of micronutrients regularly measured [23]. At the moment, there are no specific supplements designed for the KD, and concerns have been raised about the most commonly used micronutrient supplement, containing high amounts of the fat-soluble vitamins A and E [24], which are naturally high in KDs as a result of its high fat content.
A low intake of oligoelements such as zinc, selenium, and magnesium has also been reported. In a study on children on a classical KD, only 3 of the 28 micronutrients met the American dietary reference intakes [65], with zinc and magnesium particularly compromised [66]. However, Liu et al. [67] reported low levels of phosphorus and folate in otherwise normal micronutrient statuses. Close monitoring of micronutrient statuses in patients undergoing KD is therefore mandatory.
References
- Berger, M.M.; Shenkin, A.; Schweinlin, A.; Amrein, K.; Augsburger, M.; Biesalski, H.K.; Bischoff, S.C.; Casaer, M.P.; Gundogan, K.; Lepp, H.L.; et al. ESPEN micronutrient guideline. Clin. Nutr. 2022, 41, 1357–1424.
- Mehri, A. Trace Elements in Human Nutrition (II)—An Update. Int. J. Prev. Med. 2020, 11, 2.
- Shenkin, A. The key role of micronutrients. Clin. Nutr. 2006, 25, 1–13.
- Allen, L.; de Benoist, B.; Dary, O.; Hurrel, R. (Eds.) Guidelines on Food Fortification with Micronutrients; World Health Organization (WHO): Geneva, Switzerland; Food and Agricultural Organization (FAO) of the United Nations: Rome, Italy, 2006.
- Battaglia-Hsu, S.F.; Ghemrawi, R.; Coelho, D.; Dreumont, N.; Mosca, P.; Hergalant, S.; Gauchotte, G.; Sequeira, J.M.; Ndiongue, M.; Houlgatte, R.; et al. Inherited disorders of cobalamin metabolism disrupt nucleocytoplasmic transport of mRNA through impaired methylation/phosphorylation of ELAVL1/HuR. Nucleic Acids Res. 2018, 46, 7844–7857.
- Balasubramaniam, S.; Christodoulou, J.; Rahman, S. Disorders of riboflavin metabolism. J. Inherit. Metab. Dis. 2019, 42, 608–619.
- Leon-Del-Rio, A. Biotin in metabolism, gene expression, and human disease. J. Inherit. Metab. Dis. 2019, 42, 647–654.
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders affecting vitamin B(6) metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646.
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557.
- Tolomeo, M.; Nisco, A.; Leone, P.; Barile, M. Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. Int. J. Mol. Sci. 2020, 21, 5310.
- MaCdonald, A.; van Rijn, M.; Feillet, F.; Lund, A.M.; Bernstein, L.; Bosch, A.M.; Gizewska, M.; van Spronsen, F.J. Adherence issues in inherited metabolic disorders treated by low natural protein diets. Ann. Nutr. Metab. 2012, 61, 289–295.
- Singh, R.H. Nutritional management of patients with urea cycle disorders. J. Inherit. Metab. Dis. 2007, 30, 880–887.
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Belanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Gizewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162.
- Forny, P.; Horster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grunert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592.
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230.
- Merritt, J.L., 2nd; MacLeod, E.; Jurecka, A.; Hainline, B. Clinical manifestations and management of fatty acid oxidation disorders. Rev. Endocr. Metab. Disord. 2020, 21, 479–493.
- McWhorter, N.; Ndugga-Kabuye, M.K.; Puurunen, M.; Ernst, S.L. Complications of the Low Phenylalanine Diet for Patients with Phenylketonuria and the Benefits of Increased Natural Protein. Nutrients 2022, 14, 4960.
- Baumgartner, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130.
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grunewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. International clinical guideline for the management of classical galactosemia: Diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 2017, 40, 171–176.
- Cano, A.; Alcalde, C.; Belanger-Quintana, A.; Canedo-Villarroya, E.; Ceberio, L.; Chumillas-Calzada, S.; Correcher, P.; Couce, M.L.; Garcia-Arenas, D.; Gomez, I.; et al. Vitamin C and folate status in hereditary fructose intolerance. Eur. J. Clin. Nutr. 2022, 76, 1733–1739.
- Kishnani, P.S.; Goldstein, J.; Austin, S.L.; Arn, P.; Bachrach, B.; Bali, D.S.; Chung, W.K.; El-Gharbawy, A.; Brown, L.M.; Kahler, S.; et al. Diagnosis and management of glycogen storage diseases type VI and IX: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 772–789.
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, e1.
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Helen Cross, J.; Dahlin, M.G.; et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317.
- Magrath, G.; MacDonald, A.; Whitehouse, W. Dietary practices and use of the ketogenic diet in the UK. Seizure 2000, 9, 128–130.
- Lammardo, A.M.; Robert, M.; Rocha, J.C.; van Rijn, M.; Ahring, K.; Belanger-Quintana, A.; MacDonald, A.; Dokoupil, K.; Ozel, H.G.; Goyens, P.; et al. Main issues in micronutrient supplementation in phenylketonuria. Mol. Genet. Metab. 2013, 110, S1–S5.
- Singh, R.H.; Rohr, F.; Frazier, D.; Cunningham, A.; Mofidi, S.; Ogata, B.; Splett, P.L.; Moseley, K.; Huntington, K.; Acosta, P.B.; et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 121–131.
- Burlina, A.; Leuzzi, V.; Spada, M.; Carbone, M.T.; Paci, S.; Tummolo, A. The management of phenylketonuria in adult patients in Italy: A survey of six specialist metabolic centers. Curr. Med. Res. Opin. 2021, 37, 411–421.
- Barretto, J.R.; Silva, L.R.; Leite, M.E.; Boa-Sorte, N.; Pimentel, H.; Purificacao, A.C.; Carvalho, G.; Fontes, M.I.; Amorim, T. Poor zinc and selenium status in phenylketonuric children and adolescents in Brazil. Nutr. Res. 2008, 28, 208–211.
- Prochazkova, D.; Jarkovsky, J.; Vinohradska, H.; Konecna, P.; Machacova, L.; Dolezel, Z. Controlled diet in phenylketonuria and hyperphenylalaninemia may cause serum selenium deficiency in adult patients: The Czech experience. Biol. Trace Elem. Res. 2013, 154, 178–184.
- Evans, S.; Daly, A.; MacDonald, J.; Preece, M.A.; Santra, S.; Vijay, S.; Chakrapani, A.; MacDonald, A. The micronutrient status of patients with phenylketonuria on dietary treatment: An ongoing challenge. Ann. Nutr. Metab. 2014, 65, 42–48.
- De Almeida, B.N.F.; Laufer, J.A.; Mezzomo, T.R.; Shimada, N.C.; Furtado, I.H.F.; Dias, M.; Pereira, R.M. Nutritional and metabolic parameters of children and adolescents with phenylketonuria. Clin. Nutr. ESPEN 2020, 37, 44–49.
- Crujeiras, V.; Aldamiz-Echevarria, L.; Dalmau, J.; Vitoria, I.; Andrade, F.; Roca, I.; Leis, R.; Fernandez-Marmiesse, A.; Couce, M.L. Vitamin and mineral status in patients with hyperphenylalaninemia. Mol. Genet. Metab. 2015, 115, 145–150.
- Green, B.; Browne, R.; Firman, S.; Hill, M.; Rahman, Y.; Kaalund Hansen, K.; Adam, S.; Skeath, R.; Hallam, P.; Herlihy, I.; et al. Nutritional and Metabolic Characteristics of UK Adult Phenylketonuria Patients with Varying Dietary Adherence. Nutrients 2019, 11, 2459.
- Kharitonchik, L.A.; Kodentsova, V.M.; Vrzhesinskaia, O.A.; Denisova, S.N.; Spirichev, V.B. Vitamin B 6 metabolism in phenylketonuria. Vopr. Meditsinskoi Khimii 2000, 46, 81–88.
- Kastrikina, L.N.; Kopylova, N.V.; Rybakova, E.P.; Ladodo, K.S.; Churdaleva, E.V.; Spirichev, V.B. The activity of glutathione-reductase and FAD-effect as indicators of the riboflavin level in experiments and in patients with phenylketonuria. Vopr. Pitan. 1975, 5, 12–18.
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, O.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70.
- Tummolo, A.; Leone, P.; Tolomeo, M.; Solito, R.; Mattiuzzo, M.; Lepri, F.R.; Lore, T.; Cardinali, R.; De Giovanni, D.; Simonetti, S.; et al. Combined isobutyryl-CoA and multiple acyl-CoA dehydrogenase deficiency in a boy with altered riboflavin homeostasis. JIMD Rep. 2022, 63, 276–291.
- Touati, G.; Valayannopoulos, V.; Mention, K.; de Lonlay, P.; Jouvet, P.; Depondt, E.; Assoun, M.; Souberbielle, J.C.; Rabier, D.; Ogier de Baulny, H.; et al. Methylmalonic and propionic acidurias: Management without or with a few supplements of specific amino acid mixture. J. Inherit. Metab. Dis. 2006, 29, 288–298.
- Frazier, D.M.; Allgeier, C.; Homer, C.; Marriage, B.J.; Ogata, B.; Rohr, F.; Splett, P.L.; Stembridge, A.; Singh, R.H. Nutrition management guideline for maple syrup urine disease: An evidence- and consensus-based approach. Mol. Genet. Metab. 2014, 112, 210–217.
- Yannicelli, S.; Acosta, P.B.; Velazquez, A.; Bock, H.G.; Marriage, B.; Kurczynski, T.W.; Miller, M.; Korson, M.; Steiner, R.D.; Rutledge, L.; et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol. Genet. Metab. 2003, 80, 181–188.
- Pinar-Sueiro, S.; Martinez-Fernandez, R.; Lage-Medina, S.; Aldamiz-Echevarria, L.; Vecino, E. Optic neuropathy in methylmalonic acidemia: The role of neuroprotection. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S199–S203.
- Traber, G.; Baumgartner, M.R.; Schwarz, U.; Pangalu, A.; Donath, M.Y.; Landau, K. Subacute bilateral visual loss in methylmalonic acidemia. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2011, 31, 344–346.
- Matsumoto, S.; Haberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847.
- Berry, G.T.; Steiner, R.D. Long-term management of patients with urea cycle disorders. J. Pediatr. 2001, 138, S56–S60, discussion S60–S51.
- Singh, R.H. Nutrition menagement of patients with inherited disorders of urea cycle enzymes. In Nutrition Menagement of Patients with Inherited Metabolic Disorders; Acosta, P.B., Ed.; Jones and Bartlett Publishers Inc.: Sudbury, MA, USA, 2009.
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477.
- Gregersen, N.; Andresen, B.S.; Pedersen, C.B.; Olsen, R.K.; Corydon, T.J.; Bross, P. Mitochondrial fatty acid oxidation defects--remaining challenges. J. Inherit. Metab. Dis. 2008, 31, 643–657.
- Merritt, J.L., 2nd; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6, 473.
- Van Calcar, S.C.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.U.; Pendyal, S.; Wallace, L.S.; Hansen, J.G.; Stembridge, A.; et al. Nutrition management guideline for very-long chain acyl-CoA dehydrogenase deficiency (VLCAD): An evidence- and consensus-based approach. Mol. Genet. Metab. 2020, 131, 23–37.
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427.
- Goresky, C.A.; Bach, G.G.; Nadeau, B.E. On the uptake of materials by the intact liver. The transport and net removal of galactose. J. Clin. Investig. 1973, 52, 991–1009.
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Succoio, M.; Sacchettini, R.; Rossi, A.; Parenti, G.; Ruoppolo, M. Galactosemia: Biochemistry, Molecular Genetics, Newborn Screening, and Treatment. Biomolecules 2022, 12, 968.
- Ahmad, U.; Sharma, J. Fructose-1-Phosphate Aldolase Deficiency. In StatPearls; Ineligible Companies. Disclosure: Jyotsna Sharma Declares No Relevant Financial Relationships with Ineligible Companies; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Gaughan, S.; Ayres, L.; Baker, P.R., II. Hereditary Fructose Intolerance. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Chen, Y.T. Glycogen storage diseases. In The Metabolic & Molecular Basis of Inherited Diseases; Scriver, C.R., Beaudet, A.L., Sly, W.S., Vale, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 1521–1552.
- Cori, G.T.; Cori, C.F. Glucose-6-phosphatase of the liver in glycogen storage disease. J. Biol. Chem. 1952, 199, 661–667.
- Gierke, E.V. Hepato-nephro-megalia-glycogenica (Glykogenspeicherkrankheit der Leber und Nieren). Beitr. Pathol. Anat. 1929, 82, 497–513.
- Christodoulides, S.S.; Neal, E.G.; Fitzsimmons, G.; Chaffe, H.M.; Jeanes, Y.M.; Aitkenhead, H.; Cross, J.H. The effect of the classical and medium chain triglyceride ketogenic diet on vitamin and mineral levels. J. Hum. Nutr. Diet. Off. J. Br. Diet. Assoc. 2012, 25, 16–26.
- Scholl-Burgi, S.; Holler, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773.
- Mereis, M.; Wanders, R.J.A.; Schoonen, M.; Dercksen, M.; Smuts, I.; van der Westhuizen, F.H. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int. J. Biochem. Cell Biol. 2021, 132, 105899.
- Bergqvist, A.G.; Schall, J.I.; Stallings, V.A. Vitamin D status in children with intractable epilepsy, and impact of the ketogenic diet. Epilepsia 2007, 48, 66–71.
- Willmott, N.S.; Bryan, R.A. Case report: Scurvy in an epileptic child on a ketogenic diet with oral complications. Eur. Arch. Paediatr. Dent. Off. J. Eur. Acad. Paediatr. Dent. 2008, 9, 148–152.
- Hoyt, C.S.; Billson, F.A. Optic neuropathy in ketogenic diet. Br. J. Ophthalmol. 1979, 63, 191–194.
- Zupec-Kania, B.; Zupanc, M.L. Long-term management of the ketogenic diet: Seizure monitoring, nutrition, and supplementation. Epilepsia 2008, 49 (Suppl. S8), 23–26.
- Zupec-Kania, B.A.; Spellman, E. An overview of the ketogenic diet for pediatric epilepsy. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2008, 23, 589–596.
- Liu, Y.M.; Williams, S.; Basualdo-Hammond, C.; Stephens, D.; Curtis, R. A prospective study: Growth and nutritional status of children treated with the ketogenic diet. J. Am. Diet. Assoc. 2003, 103, 707–712.
More
Information
Subjects:
Nutrition & Dietetics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
686
Revisions:
2 times
(View History)
Update Date:
19 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No