Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Cristina Hernández-Ramírez | -- | 3531 | 2023-12-13 19:41:34 | | | |
| 2 | Mona Zou | Meta information modification | 3531 | 2023-12-14 09:24:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Torres-Morán, M.; Franco-Álvarez, A.L.; Rebollar-Vega, R.G.; Hernández-Ramírez, L.C. Somatic Genetic Variation in Pituitary Neuroendocrine Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/52709 (accessed on 24 June 2026).
Torres-Morán M, Franco-Álvarez AL, Rebollar-Vega RG, Hernández-Ramírez LC. Somatic Genetic Variation in Pituitary Neuroendocrine Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/52709. Accessed June 24, 2026.
Torres-Morán, Mariana, Alexa L. Franco-Álvarez, Rosa G. Rebollar-Vega, Laura C. Hernández-Ramírez. "Somatic Genetic Variation in Pituitary Neuroendocrine Tumors" Encyclopedia, https://encyclopedia.pub/entry/52709 (accessed June 24, 2026).
Torres-Morán, M., Franco-Álvarez, A.L., Rebollar-Vega, R.G., & Hernández-Ramírez, L.C. (2023, December 13). Somatic Genetic Variation in Pituitary Neuroendocrine Tumors. In Encyclopedia. https://encyclopedia.pub/entry/52709
Torres-Morán, Mariana, et al. "Somatic Genetic Variation in Pituitary Neuroendocrine Tumors." Encyclopedia. Web. 13 December, 2023.
Copy Citation
Mutational hotspots have gained importance as oncological biomarkers in recent years because of their potential as predictors of clinical outcomes and/or therapeutic targets. In addition, they are easily detectable in clinical samples via Sanger or next-generation sequencing (NGS). The role of these genetic defects is less clear in pituitary neuroendocrine tumors (PitNETs), even though the most common genetic drivers of these neoplasms are located within mutational hotspots. Indeed, hotspots in six different genes are of particular importance in this context. Two of them, USP48 and SF3B1, represent very recent and infrequent genetic associations; thus, their clinical relevance remains unclear. For two other genes, GNAS and USP8, discrepancies exist among studies regarding their associated phenotypes. Finally, the phenotypes associated with BRAF and DICER1 are well defined in other settings, but not yet in sporadic PitNETs. Additional studies are required to assess the potential of these molecular alterations as druggable targets in PitNETs.
genetic driver
mutational hotspot
pituitary neuroendocrine tumor
somatic variant
druggable target
1. BRAF
Protein kinase is the most frequently shared domain among cancer-associated proteins, therefore representing a particularly attractive therapeutic target [1]. The isoforms A, B, and C of the highly conserved serine/threonine protein kinase rapidly accelerated fibrosarcoma (RAF) proteins, encoded in humans by three different genes, are among such proteins. C-RAF (also known as RAF-1) was first described in 1985, while A-RAF was discovered in 1986, and B-RAF in 1988 [2][3][4]. The latter is encoded by BRAF (7q34, RefSeq NM_001354609.2), a proto-oncogene with preferential expression in neural tissues, and is the most potent activator of the RAS-GTPase (RAS)-RAF-MAPK and ERK kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway (RAS-RAF-MEK-ERK pathway) [5][6][7] (Figure 1). This phosphorylation cascade is involved in the physiological regulation of cellular processes such as proliferation, survival, differentiation, apoptosis, and motility [6].
Germline activating variants affecting either BRAF or other members of the RAS-RAF-MEK-ERK pathway are associated with a group of developmental syndromes collectively known as RASopathies [8]. In contrast, the upregulation of this pathway via various mechanisms contributes to tumorigenesis in one-third of human cancers [9]. Specifically, somatic missense activating variants in the glycine-rich loop or the activation segment of the BRAF catalytic domain occur in about 7% of all cancers. At least 90% of such cases, however, are explained by a single defect: c.1799T>A, p.V600E [10][11]. This variant is found in two-thirds of malignant melanomas and papillary thyroid carcinomas (PTCs) and less frequently in colorectal, ovarian, and other types of cancer [10][11][12].
The phosphorylation of residues T599 and S602 (UniProt P15056), which flank the variant, is required for BRAF to be recruited to the cell membrane and folded into its active conformation. The p.V600E change destabilizes the inactive conformation of BRAF and promotes its active state, thereby acting as a phosphomimetic [13]. This way, BRAF p.V600E results in an abnormally active RAS-independent kinase that induces cell proliferation and transformation in vitro and in vivo [10][14][15]. Indeed, BRAF variants seem to be mutually exclusive with oncogenic RAS defects [10]. In addition to the phosphorylation of the well-known downstream effectors MEK1/2, BRAF p.V600E activates NFKB and prevents apoptosis [14][16]. In colorectal cancer, BRAF p.V600E has been associated with poor clinical prognosis and chemoresistance, increased microsatellite instability, and a higher mutational load [17]. In addition, quantitation of BRAF p.V600E by droplet digital polymerase chain reaction (ddPCR) has been used as a marker for measurable residual disease in hairy cell leukemia [18].
Individual case reports of PCP treatment with drugs targeting BRAF and/or other RAS-RAF-MEK-ERK components have shown encouraging results [19]. Very recently, a phase 2 clinical trial of combined vemurafenib/cobimetinib treatment in PCP showed a response in 94% of participants, with a median tumor reduction of 91% at 22 months, for progression-free survival of 87 and 58% at 12 and 24 months, respectively [20]. In contrast, BRAF inhibitors have not been evaluated as therapeutic agents for CD in clinical trials. There are, however, three single-case reports of BRAF p.V600E positive posterior pituitary tumors (two with confirmed NKX2-1-expression) treated with dabrafenib, either alone [21] or combined with cobimetinib [22] or trametinib [23]. All tumors had recurred after one or more surgeries plus radiotherapy. One patient developed stable disease [23] and two experienced significant tumor regression [21], although the combined therapy resulted in dermatological toxicity.
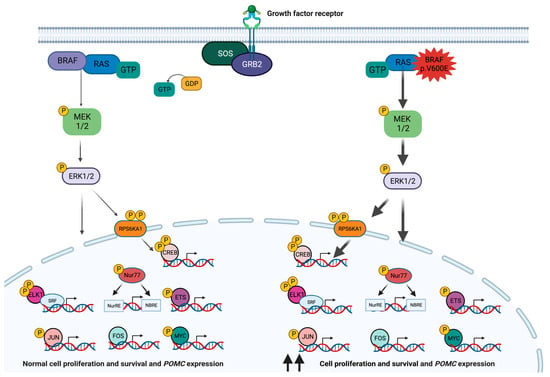
Figure 1. The RAS-RAF-MEK-ERK signaling pathway in corticotroph cells. Under physiological conditions, this pathway is activated in response to the interaction of extracellular ligands such as growth factors, hormones, or cytokines with a tyrosine kinase receptor. The receptor-like growth factor receptor-binding protein 2 (GRB2) binds to the activated receptor and interacts with the proline-rich sequence at the C-terminus of the son of sevenless (SOS) protein to form the receptor-GRB2-SOS complex, which in turn promotes the GTP-mediated activation of RAS. Activated RAS protein binds to and recruits BRAF to the inner side of the cell membrane, where it is phosphorylated by tyrosine kinases. The C-terminal catalytic domain of BRAF interacts with and phosphorylates MEK1 and 2 into their catalytic VIII subregion. In turn, MEK1 and 2 phosphorylate and thus activate ERK1 and 2 (also known as mitogen-activated protein kinases (MAPK) 3 and 1). In addition to phosphorylating cytoplasmic targets, active ERK1 and 2 enter the nucleus and phosphorylate multiple transcription factors, such as ELK1, ETS, FOS, JUN, and MYC, thereby inducing the expression of their target genes. Via the phosphorylation of RPS6KA1, ERK1 and 2 also activate the transcription factor cAMP response element-binding protein (CREB). The activation of this pathway leads to tissue-specific molecular consequences, although in the pituitary gland and in many other tissues it results in increased cell proliferation and survival [6][11][24][25]. In corticotroph cells, this pathway also activates POMC transcription, although the membrane receptor triggering this response in physiological conditions and in corticotropinomas remains unclear [26]. The BRAF p.V600E variant leads to the overactivation of this signaling pathway.
2. GNAS
At least ~100 human genes are subjected to genomic imprinting, an epigenetic mechanism that controls gene expression in a parent-of-origin and tissue-specific manner [27]. Using differentially imprinted promoters, one of these genes, GNAS (locus of the GNAS complex, 20q13.32), ultimately translates into multiple proteins, namely XLαs, ALEX, NESP55, and Gsα [28][29]. The latter, encoded by a 13-exon reference transcript (NM_000516.7), accounts for the 394-amino-acid α subunit of the heterotrimeric stimulating G protein (P63092-1) [30]. Gsα is translated from the maternal allele in the pituitary, thyroid, and gonads, but depends on biallelic expression in other tissues [31].
At the molecular level, guanine nucleotide-binding proteins (G proteins) function as information transducers between the cell-membrane-bound G-protein-coupled receptors (GPCRs) and their effectors, thereby regulating the production of second messengers [32]. G proteins are composed of α, β, and γ subunits (encoded by different genes) and form a complex that binds GPCRs [33]. Gsα is made of a C-terminal RAS-like guanosine triphosphatase (GTPase) that also functions as an interaction site for the β and γ subunits and an N-terminal helicoidal domain [34]. A nucleotide binding cleft exists in between those two domains, which binds guanosine diphosphate (GDP) while the GPCR is inactive. Following GPCR activation through ligand binding, Gsα exchanges GDP for guanosine triphosphate (GTP) and dissociates from the βγ dimer and the receptor, thereby allowing for the GNAS-dependent activation of adenylyl cyclases (ACs) [35][36]. ACs in turn catalyze the synthesis of cyclic 3′,5′-adenosine monophosphate (cAMP), which then activates downstream signaling pathways [33]. This activation cycle is negatively regulated by the intrinsic GTPase activity of Gsα, which prevents the continued activation of downstream effectors [35] (Figure 2). The effects of multiple hormones greatly depend on cAMP, and the specificity of the cellular responses elicited by this second messenger is determined in a tissue-specific manner [37].
Missense GNAS variants affecting residues R201 (namely p.R201C, p.R201S, and p.R201H), and G227 (p.G227R, p.G227L, and p.G227K) of GNAS have been described in endocrine tumors and other human neoplasms. They have been found as somatic changes in somatotropinomas (4.4–59.5%), non-functioning PitNETs (7–10%), corticotropinomas (6%), autonomous thyroid adenomas (5%) and thyroid cancer (13% of PTC and up to 4% of follicular tumors), and occasionally, in ovarian and testicular Leydig cell tumors, prolactinomas, adrenocortical adenomas, pheochromocytomas, paragangliomas, parathyroid adenomas, and in patients with multiple endocrine tumors [38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66]. These variants have also been found in non-endocrine malignant neoplasms, such as pancreatic, colorectal, and lung adenocarcinomas, as well as in hepatocellular carcinomas [67][68][69][70].
GNAS variants also underlie the McCune–Albright syndrome (MAS, MIM #174800), a rare condition with sporadic presentation characterized by genetic mosaicism due to early postzygotic GNAS hotspot defects [71][72]. The diagnosis is established in the presence of two or more of the classic MAS features: polyostotic fibrous dysplasia, café-au-lait skin spots, and endocrine hyperfunction (gonadotropin-independent precocious puberty, hyperthyroidism, early-onset Cushing’s syndrome, and PitNETs, usually GH or GH and prolactin-secreting, among others) [73]. Ninety-five percent of MAS cases are due to variants in R201, while only 5% are caused by variants in Q227 [74][75][76]. The phenotype is determined by genomic imprinting and the disease severity correlates with the degree of mosaicism, meaning that the clinical presentation depends on the time of appearance of the GNAS variant during embryogenesis [77].
GNAS hotspot variants cause the loss of protein function that results in increased activity of the cAMP signaling pathway, by (1) stabilizing Gsα in its active conformation, thereby mimicking the effect of extracellular growth factors by stimulating ACs, and (2) inhibiting GTPase activity and causing a constitutive activation of ACs [38][78]. For these reasons, these GNAS defects are often referred to as activating variants or gsp oncogene [38]. Restoring the GTPase activity of GNAS is an attractive therapeutic target, although drugs with this specific effect have not been reported yet. In contrast, non-hotspot loss-of-function (LOF) GNAS variants cause Albright’s hereditary osteodystrophy [79].
The clinical consequences of GNAS variants have been thoroughly studied in somatotropinomas. Some studies have defined a particular GNAS-associated phenotype, with patients usually being older and presenting significantly smaller tumors associated with low serum GH or IGF1 levels [39][57][64][65]. Other studies have described GNAS-driven tumors as having a slow growth rate and a better response to pharmacological or surgical treatment compared with wild-type tumors [64][66][80]. These tumors are usually of the densely granulated subtype at the histopathological examination [81]. Differences in age, sex, and other clinical characteristics have been suggested by some studies [57][82][83]. At the molecular level, GNAS hotspot variants define a distinctive subgroup of somatotropinomas that display hypomethylation, limited chromosomal alterations, and activation of the GPCR pathway, although results vary among studies [66][84][85]. In both sporadic and MAS-related somatotropinomas, GNAS variants almost always affect the maternal allele [86]. While wild-type somatotropinomas often display relaxation of the paternal imprinting, this phenomenon is infrequent in tumors carrying GNAS variants [65][87][88]. The relaxation of GNAS imprinting correlates with lower GNAS, SSTR2, and AIP expression, suggesting a possibly reduced response to somatostatin receptor ligands [65].
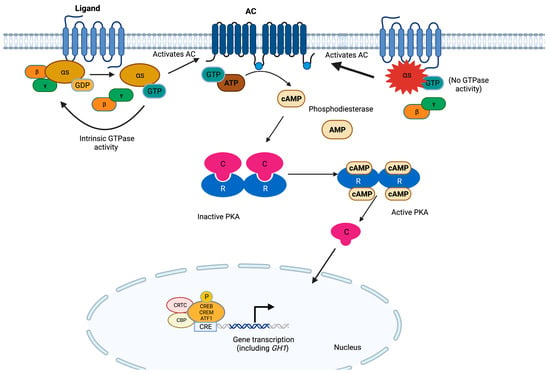
Figure 2. The cAMP pathway in somatotroph cells. G proteins are composed of three subunits, and the α subunit contains high-affinity binding sites for guanine nucleotides. The GDP-bound form binds tightly to βγ and is inactive, whereas the GTP-bound form dissociates from βγ and is the active form. GPCRs cause the activation of G proteins by facilitating the exchange of GTP for GDP on the α subunit, which in turn activates ACs. These enzymes use ATP as a substrate to produce cAMP. The latter binds to the regulatory subunits (R) of PKA, allowing for the release of the catalytic subunits (C). Active PKA catalyzes the serine/threonine phosphorylation of target molecules, including the transcription factors CREB, CRE modulator (CREM), and activating transcription factor 1 (ATF1). In complex with co-activators such as CREB-binding protein (CBP) and members of the cAMP-regulated transcriptional co-activators (CRTC), these transcription factors bind the 8 bp palindromic sequence known as cAMP response element (CRE) in the promoter region of target genes to increase their transcription. In somatotrophs, the GH-releasing hormone receptor (GHRHR) is the main GPCR activating this pathway, promoting both cell proliferation and GH transcription [33][34][35][36][89]. GNAS hotspot variants result in the constitutive activation of this pathway.
4. DICER1
The DICER1 syndrome (MIM #601200) is an autosomal dominant condition of tumor predisposition that encompasses otherwise infrequent dysembryonic tumors, such as pleuropulmonary blastoma (PPB), cystic nephroma (CN), ovarian sex cord stromal tumor, nasal chondromesenchymal hamartoma, ciliary body and cerebral medulloepitheliomas, anaplastic kidney sarcoma, pineoblastoma, embryonal rhabdomyosarcoma (ERMS), and pituitary blastoma (PitB) [90]. Other associated neoplasms are Wilms tumor (WT), juvenile hamartomatous intestinal polyps, and differentiated thyroid carcinoma, as well as benign lesions such as multinodular goiter and pulmonary cysts.
This syndrome presents usually at an early age and occasionally in young adults and is caused in most cases by germline heterozygous LOF DICER1 (14q32.13) variants that appear de novo in 10–20% of cases [91][92][93][94]. Ten percent of cases are due to somatic mosaicism for DICER1 variants, which has been associated with earlier disease onset, more DICER1-associated tumors, and a distinctive presentation known as GLOW syndrome (global developmental delay, lung cysts, overgrowth, and Wilms tumor) [93][95][96].
The 29-exon DICER1 canonical transcript (NM_030621.4) encodes a widely expressed 1922 amino acid cytoplasmic enzyme (Q9UPY3-1) composed, from N- to C-terminal, of a helicase domain, a domain of unknown function (DUF283), a platform domain, a P-element-induced whimpy tested (PIWI)-Argonaute (AGO)-Zwille (PAZ) domain, a connector domain, the class 3 ribonuclease (RNase III) a and b domains, and a double-stranded RNA (dsRNA)-binding domain [97]. DICER1 plays a crucial role in the processing of small RNAs, which are the RNA species involved in gene silencing. It first cleaves pre-miRNAs and long dsRNA substrates into mature microRNAs (miRNAs) and small interfering RNAs (siRNAs), respectively [98][99]. Then, DICER1 participates in the loading of siRNAs and miRNAs onto the RNA-induced silencing complex (RISC), composed of DICER1, an AGO protein, and the RISC-loading complex subunit transactivating response RNA-binding protein (TARBP2) [100]. The AGO protein selects a strand of the small RNA as a guide, which in turn directs the small RNA-bound RISC complex toward complementary messenger RNA (mRNA) sequences. The mRNA targets are then either cleaved by AGO (RNA interference) or translationally repressed and directed to degradation (miRNA-mediated gene silencing); the latter mechanism predominates in mammalian cells [101] (Figure 3).
Most individuals carrying germline DICER1 variants also harbor somatic second hits, which in most cases are missense changes and rarely loss of heterozygosity (LOH) [96]. Moreover, somatic deleterious DICER1 variants have been reported in the presence or absence of germline defects in patients with PPB, CN, WT, non-epithelial ovarian tumors, cervical ERMS, PitB, prostate carcinoma, pineoblastoma, differentiated thyroid carcinoma, and testicular germ cell tumors [102][103][104][105][106][107][108][109][110][111][112]. Different to germline variants, which are usually truncating and are not clustered in hotspots, most mosaic and somatic variants occurring isolated or as second hits are missense and located within the RNase IIIb domain [96][109][113].
Nineteen out of the twenty PitBs genotyped so far were due to LOF DICER1 variants, although it is not clear if any cases were caused by somatic defects [94][114][115][116]. These tumors usually affect neonates or infants, but one case diagnosed in childhood and one presenting in young adulthood have been reported [114][115][116]. These extremely rare and poorly differentiated anterior pituitary neoplasms with a so-called oncofetal molecular signature usually express ACTH and may present clinically silently or as CD [109][117][118]. Nine of these patients died during infancy or childhood due to tumor-related complications [115][116]. Because PitB is considered a pathognomonic lesion of the DICER1 syndrome, its diagnosis should prompt germline DICER1 screening and genetic counseling [94].
RNAse IIIb variants affect metal ion binding and adjacent amino acids, specifically 1705, 1709, 1809, 1810, or 1813, which are therefore considered missense hotspots [90][99]. Second somatic variants outside the hotspot as well as LOH have also been described in patients with somatic mosaicism for RNAse IIIb variants [93][112]. The abnormal RNase IIIb cleaves 5′-derived miRNAs from the pre-miRNA hairpin loops inefficiently, causing retention of pre-miRNA loop sequences and leading to reduced expression of 5′-derived mature miRNAs and predominance of 3′-derived pre-miRNAs [112]. The oncogenic capacity of the biased pre-miRNA repertoire seems to depend on the cellular and developmental setting [113].
Aside from its role as a tumor driver, reduced DICER1 expression due to haploinsufficiency or other mechanisms correlates with bad outcomes in multiple types of cancer [99]. In these tumors, unprocessed pre-miRNAs are degraded by the endonuclease complex TSN-TSNAX. Pharmacological or shRNA-mediated inhibition of this complex facilitates the restoration of miRNA levels by DICER1 in vitro, making it a potential therapeutic target [119][120]. This strategy, however, has not yet been explored in tumors carrying DICER1 hotspot variants.
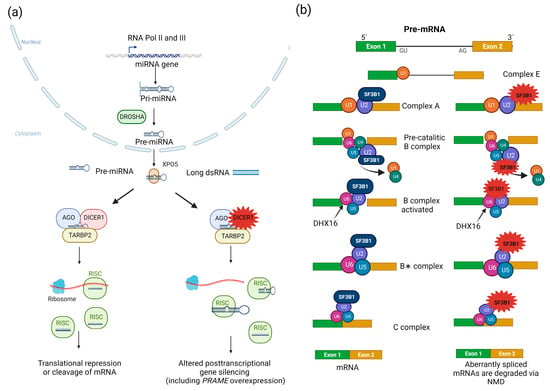
Figure 3. RNA processing pathways involved in PitNETs. (a) Biogenesis of small RNAs. In the nucleus, RNA polymerases II and III (RNA Pol II and III) generate primary miRNA transcripts (pri-miRNAs) from miRNA-encoding genes, which are then processed by the microprocessor complex, including the DROSHA RNaseIII. This initial step renders ~60-nucleotide-long hairpin-folded pre-miRNAs, which are in turn exported to the cytoplasm via exportin 5 (XPO5)/Ran-GTP. In the cytoplasm, DICER1 cleaves pre-miRNAs and long dsRNAs into mature miRNAs and siRNAs, respectively, both of which are 20–22 nucleotide-long double-stranded RNAs. The DICER1-dsRNA complex is then bound by a member of the AGO protein family (AGO2 is the best characterized of them) and TARBP2 to form the RISC-loading complex. This complex in turn loads dsRNAs into the RISC, which is required to produce single-stranded small RNAs that serve as a guide to recognize complementary RNA sequences (located in the 3′ untranslated region of mRNAs). The small RNA-loaded RISC can either block translation and promote degradation or directly cleave (via AGO proteins) target mRNAs. Additional roles for DICER1 in the responses to DNA damage (nuclear) and viral infections (cytoplasmic) have recently been described. In PitBs, this abnormal repertoire of small RNAs results in PRAME dysregulation, among other transcriptional alterations [99][101][121][122][123]. DICER1 variants result in abnormal processing of small RNAs, thereby impairing their ability to regulate gene expression. (b) Processing of mRNAs by the spliceosome. The spliceosome is a large complex of snRNPs and other proteins that carries out the removal of introns and the ligation of exons from mRNA precursors (pre-mRNAs), rendering mature mRNAs. Two types of spliceosomes, U2-dependent and U12-dependent, are recognized in eukaryotes, the former being the predominant one. The U2-dependent spliceosome is composed of U1, U2, U5, and U4/U6 snRNP, as well as other proteins. This process beings when the U1 snRNP binds to the 5′ SS to form the E complex. Then, the non-ribonucleoprotein complex components SF1, U2AF2, and U2AF1 bind the BS (18–40 nucleotides upstream from the 3′ SS), the polypyrimidine tract (a sequence immediately downstream from the BS), and an AG dinucleotide at the intron-exon junction, respectively. The U2 snRNP in turn replaces SF1, forming the A complex, and the U5, and U4/U6 snRNPs are then recruited to form the precatalytic B complex. Rearrangements in RNA–RNA and RNA–protein interactions ultimately lead to dissociation of the U1 and U4 snRNPs, thereby producing the active B complex. The latter is activated by the pre-mRNA-splicing factor ATP-dependent RNA helicase DHX16, thereby generating the B∗ complex, which catalyzes the first step of splicing. The C complex is then formed, triggering the second step of splicing. Finally, the spliceosome is removed and recycled. SF3B1 hotspot variants lead to the use of cryptic pre-mRNA 3′ SSs, and aberrantly spliced mRNAs are degraded via NMD [124][125][126][127][128][129][130]. The repertoire of aberrantly spliced mRNAs involved in lactotroph tumorigenesis remains unknown.
3. SF3B1
Using genome sequencing in 21 patients and targeted genotyping by ddPCR in the rest, a recurrent missense somatic variant (c.1874G>A, p.R625H) in the splicing factor 3B subunit 1 gene (SF3B1, 2q33.1, NM_012433.4) was identified in 20% of prolactinomas of a single cohort of 227 cases [131]. When 154 PitNETs of other types were tested, this variant was only found in 6% of cases, all of them staining positive for prolactin. Individuals carrying SF3B1 p.R625H displayed significantly higher prolactin levels and a shorter progression-free survival, compared with SF3B1 wild-type cases. A recent Sanger sequencing-based study identified the same variant and an additional missense variant in the same residue (c.1873C>T, p.R625C) in 7 out of 282 prolactinomas analyzed (2.5%) [132]. Interestingly, 50% of metastatic prolactinomas carried SF3B1 hotspot defects. In line with the earlier findings, SF3B1 variants were associated with a larger tumor size and increased mortality, but also with a higher Ki67 index and a need for more therapeutic interventions.
SF3B1 encodes a component of the U2 small nuclear ribonucleoprotein (snRNP) complex and is therefore a component of the pre-mRNA splicing machinery. SF3B1 is involved in 3′ acceptor splice site (SS) recognition, as well as in recruiting other U2 snRNP subunits to the branch point (BP) of pre-mRNAs via interaction with the BP and U2AF2 [133] (Figure 3). The canonical form of SF3B1 (O75533-1) is a 1304-amino-acid protein containing an unstructured N-terminal region, while the C-terminal two-thirds of the protein constitute a huntingtin, elongation factor 3, regulatory A subunit of protein phosphatase 2A, and TOR1 (HEAT) domain, composed of 20 tandem repeats [124].
Recurrent somatic variants in hotspots within the fifth and ninth HEAT repeats have been found in myelodysplastic syndrome, chronic myelomonocytic leukemia, acute myeloid leukemia, myeloproliferative neoplasms, primary myelofibrosis, chronic lymphocytic leukemia, breast cancer, pancreatic ductal adenocarcinoma, uveal, mucosal, and cutaneous melanoma, and prostate cancer [125][134][135][136][137][138][139][140][141]. Aberrant splicing is a well-known tumorigenic mechanism, and indeed, abnormal splicing patterns have been demonstrated in neoplasms carrying SF3B1 hotspot variants in some [134][135], although not all studies [139]. In prolactinomas, p.R625H (in the fifth HEAT repeat) leads to aberrant splicing of estrogen-related receptor gamma (ESRRG) mRNA, resulting in stronger interaction with the pituitary-specific positive transcription factor 1 (POU1F1) and excessive prolactin secretion [131]. This variant also causes aberrant splicing and downregulation of DLG1 in human prolactinomas and rat somatotropinoma GH3 cells. In the latter, the variant causes an epithelial–mesenchymal transition phenotype [142].
References
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158.
- Bonner, T.I.; Kerby, S.B.; Sutrave, P.; Gunnell, M.A.; Mark, G.; Rapp, U.R. Structure and biological activity of human homologs of the raf/mil oncogene. Mol. Cell Biol. 1985, 5, 1400–1407.
- Huebner, K.; ar-Rushdi, A.; Griffin, C.A.; Isobe, M.; Kozak, C.; Emanuel, B.S.; Nagarajan, L.; Cleveland, J.L.; Bonner, T.I.; Goldsborough, M.D.; et al. Actively transcribed genes in the raf oncogene group, located on the X chromosome in mouse and human. Proc. Natl. Acad. Sci. USA 1986, 83, 3934–3938.
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. B-raf, a new member of the raf family, is activated by DNA rearrangement. Mol. Cell Biol. 1988, 8, 2651–2654.
- Sithanandam, G.; Druck, T.; Cannizzaro, L.A.; Leuzzi, G.; Huebner, K.; Rapp, U.R. B-raf and a B-raf pseudogene are located on 7q in man. Oncogene 1992, 7, 795–799.
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885.
- Gualtieri, A.; Kyprianou, N.; Gregory, L.C.; Vignola, M.L.; Nicholson, J.G.; Tan, R.; Inoue, S.I.; Scagliotti, V.; Casado, P.; Blackburn, J.; et al. Activating mutations in BRAF disrupt the hypothalamo-pituitary axis leading to hypopituitarism in mice and humans. Nat. Commun. 2021, 12, 2028.
- Hebron, K.E.; Hernandez, E.R.; Yohe, M.E. The RASopathies: From pathogenetics to therapeutics. Dis. Model Mech. 2022, 15, dmm049107.
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319.
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262.
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867.
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003, 63, 8132–8137.
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342.
- Hingorani, S.R.; Jacobetz, M.A.; Robertson, G.P.; Herlyn, M.; Tuveson, D.A. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003, 63, 5198–5202.
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: A review of treatment options and evidence-based guidelines. Ann. Oncol. 2021, 32, 959–967.
- Broccoli, A.; Terragna, C.; Nanni, L.; Martello, M.; Armuzzi, S.; Agostinelli, C.; Morigi, A.; Casadei, B.; Pellegrini, C.; Stefoni, V.; et al. Droplet digital polymerase chain reaction for the assessment of disease burden in hairy cell leukemia. Hematol. Oncol. 2022, 40, 57–62.
- Juratli, T.A.; Jones, P.S.; Wang, N.; Subramanian, M.; Aylwin, S.J.B.; Odia, Y.; Rostami, E.; Gudjonsson, O.; Shaw, B.L.; Cahill, D.P.; et al. Targeted treatment of papillary craniopharyngiomas harboring BRAF V600E mutations. Cancer 2019, 125, 2910–2914.
- Brastianos, P.K.; Twohy, E.; Geyer, S.; Gerstner, E.R.; Kaufmann, T.J.; Tabrizi, S.; Kabat, B.; Thierauf, J.; Ruff, M.W.; Bota, D.A.; et al. BRAF-MEK inhibition in newly diagnosed papillary craniopharyngiomas. N. Engl. J. Med. 2023, 389, 118–126.
- Grenier-Chartrand, F.; Barrit, S.; Racu, M.L.; Luce, S.; Spitaels, J.; Sadeghi-Meibodi, N.; Lebrun, L.; Salmon, I.; Lefranc, F.; De Witte, O. Dabrafenib monotherapy for a recurrent BRAFV600E-mutated TTF-1-positive posterior pituitary tumor. Acta. Neurochir. 2022, 164, 737–742.
- Sollfrank, L.; Lettmaier, S.; Erdmann, M.; Uslu, U. Panniculitis under successful targeted inhibition of the MAPK/ERK signaling pathway in a patient with BRAF V600E-mutated spindle cell oncocytoma of the pituitary gland. Anticancer Res. 2019, 39, 3955–3959.
- Dawoud, F.M.; Naylor, R.M.; Giannini, C.; Swanson, A.A.; Meyer, F.B.; Uhm, J.H. TTF-1 positive posterior pituitary tumor: Limitations of current treatment and potential new hope in BRAF V600E mutation variants. Clin. Neurol. Neurosurg. 2020, 196, 106059.
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293.
- Derwich, A.; Sykutera, M.; Brominska, B.; Rubis, B.; Ruchala, M.; Sawicka-Gutaj, N. The role of activation of PI3K/AKT/mTOR and RAF/MEK/ERK pathways in aggressive pituitary adenomas-new potential therapeutic approach—A systematic review. Int. J. Mol. Sci. 2023, 24, 10952.
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171.
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Aspects Med. 2013, 34, 826–840.
- Hayward, B.E.; Moran, V.; Strain, L.; Bonthron, D.T. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 15475–15480.
- Hayward, B.E.; Bonthron, D.T. An imprinted antisense transcript at the human GNAS1 locus. Hum. Mol. Genet 2000, 9, 835–841.
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic. Acids Res. 2023, 51, D523–D531.
- Mantovani, G.; Ballare, E.; Giammona, E.; Beck-Peccoz, P.; Spada, A. The gsalpha gene: Predominant maternal origin of transcription in human thyroid gland and gonads. J. Clin. Endocrinol. Metab. 2002, 87, 4736–4740.
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, signaling, and physiological functions of G-proteins. J. Mol. Biol. 2016, 428, 3850–3868.
- Peverelli, E.; Mantovani, G.; Lania, A.G.; Spada, A. cAMP in the pituitary: An old messenger for multiple signals. J. Mol. Endocrinol. 2014, 52, R67–R77.
- Duc, N.M.; Kim, H.R.; Chung, K.Y. Structural mechanism of G protein activation by G protein-coupled receptor. Eur. J. Pharmacol. 2015, 763, 214–222.
- Lambright, D.G.; Noel, J.P.; Hamm, H.E.; Sigler, P.B. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature 1994, 369, 621–628.
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell. Biol. 2002, 3, 639–650.
- Hernandez-Ramirez, L.C.; Trivellin, G.; Stratakis, C.A. Cyclic 3’,5’-adenosine monophosphate (cAMP) signaling in the anterior pituitary gland in health and disease. Mol. Cell Endocrinol. 2018, 463, 72–86.
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696.
- Landis, C.A.; Harsh, G.; Lyons, J.; Davis, R.L.; McCormick, F.; Bourne, H.R. Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J. Clin. Endocrinol. Metab. 1990, 71, 1416–1420.
- Lyons, J.; Landis, C.A.; Harsh, G.; Vallar, L.; Grunewald, K.; Feichtinger, H.; Duh, Q.Y.; Clark, O.H.; Kawasaki, E.; Bourne, H.R. Two G protein oncogenes in human endocrine tumors. Science 1990, 249, 655–659.
- Suarez, H.G.; du Villard, J.A.; Caillou, B.; Schlumberger, M.; Parmentier, C.; Monier, R. gsp mutations in human thyroid tumours. Oncogene 1991, 6, 677–679.
- Spada, A.; Vallar, L. G-protein oncogenes in acromegaly. Horm. Res. 1992, 38, 90–93.
- Hosoi, E.; Yokogoshi, Y.; Hosoi, E.; Horie, H.; Sano, T.; Yamada, S.; Saito, S. Analysis of the Gs alpha gene in growth hormone-secreting pituitary adenomas by the polymerase chain reaction-direct sequencing method using paraffin-embedded tissues. Acta Endocrinol. 1993, 129, 301–306.
- Parma, J.; Duprez, L.; Van Sande, J.; Cochaux, P.; Gervy, C.; Mockel, J.; Dumont, J.; Vassart, G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993, 365, 649–651.
- Tordjman, K.; Stern, N.; Ouaknine, G.; Yossiphov, Y.; Razon, N.; Nordenskjold, M.; Friedman, E. Activating mutations of the Gs alpha-gene in nonfunctioning pituitary tumors. J. Clin. Endocrinol. Metab. 1993, 77, 765–769.
- Yoshimoto, K.; Iwahana, H.; Fukuda, A.; Sano, T.; Itakura, M. Rare mutations of the Gs alpha subunit gene in human endocrine tumors. Mutation detection by polymerase chain reaction-primer-introduced restriction analysis. Cancer 1993, 72, 1386–1393.
- Michiels, F.M.; Caillou, B.; Talbot, M.; Dessarps-Freichey, F.; Maunoury, M.T.; Schlumberger, M.; Mercken, L.; Monier, R.; Feunteun, J. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 10488–10492.
- Williamson, E.A.; Ince, P.G.; Harrison, D.; Kendall-Taylor, P.; Harris, P.E. G-protein mutations in human pituitary adrenocorticotrophic hormone-secreting adenomas. Eur. J. Clin. Invest 1995, 25, 128–131.
- Williamson, E.A.; Johnson, S.J.; Foster, S.; Kendall-Taylor, P.; Harris, P.E. G protein gene mutations in patients with multiple endocrinopathies. J. Clin. Endocrinol. Metab. 1995, 80, 1702–1705.
- Yang, I.M.; Woo, J.T.; Kim, S.W.; Kim, J.W.; Kim, Y.S.; Choi, Y.K. Characteristics of acromegalic patients with a good response to octreotide, a somatostatin analogue. Clin. Endocrinol. 1995, 42, 295–301.
- Fragoso, M.C.B.V.; Latronico, C.; Carvalho, F.M.; Zerbini, M.C.N.; Marcondes, J.A.M.; Araujo, L.M.B.; Lando, V.S.; Frazzatto, E.T.; Mendonca, B.B.; Villares, S.M.F. Activating mutation of the stimulatory G protein (gsp) as a putative cause of ovarian and testicular human stromal Leydig cell tumors. J. Clin. Endocr. Metab. 1998, 83, 2074–2078.
- Shi, Y.; Tang, D.; Deng, J.; Su, C. Detection of gsp oncogene in growth hormone-secreting pituitary adenomas and the study of clinical characteristics of acromegalic patients with gsp-positive pituitary tumors. Chin. Med. J. 1998, 111, 891–894.
- Buchfelder, M.; Fahlbusch, R.; Merz, T.; Symowski, H.; Adams, E.F. Clinical correlates in acromegalic patients with pituitary tumors expressing GSP oncogenes. Pituitary 1999, 1, 181–185.
- Persani, L.; Lania, A.; Alberti, L.; Romoli, R.; Mantovani, G.; Filetti, S.; Spada, A.; Conti, M. Induction of specific phosphodiesterase isoforms by constitutive activation of the cAMP pathway in autonomous thyroid adenomas. J. Clin. Endocrinol. Metab. 2000, 85, 2872–2878.
- Krohn, K.; Paschke, R. Somatic mutations in thyroid nodular disease. Mol. Genet. Metab. 2002, 75, 202–208.
- Mendoza, V.; Sosa, E.; Espinosa-de-los-Monteros, A.L.; Salcedo, M.; Guinto, G.; Cheng, S.; Sandoval, C.; Mercado, M. GSPalpha mutations in Mexican patients with acromegaly: Potential impact on long term prognosis. Growth Horm. IGF. Res. 2005, 15, 28–32.
- Freda, P.U.; Chung, W.K.; Matsuoka, N.; Walsh, J.E.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.N.; Post, K.D. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282.
- Taboada, G.F.; Tabet, A.L.; Naves, L.A.; de Carvalho, D.P.; Gadelha, M.R. Prevalence of gsp oncogene in somatotropinomas and clinically non-functioning pituitary adenomas: Our experience. Pituitary 2009, 12, 165–169.
- Lu, J.Y.; Hung, P.J.; Chen, P.L.; Yen, R.F.; Kuo, K.T.; Yang, T.L.; Wang, C.Y.; Chang, T.C.; Huang, T.S.; Chang, C.C. Follicular thyroid carcinoma with NRAS Q61K and GNAS R201H mutations that had a good (131)I treatment response. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 150067.
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372.
- Ritvonen, E.; Pitkanen, E.; Karppinen, A.; Vehkavaara, S.; Demir, H.; Paetau, A.; Schalin-Jantti, C.; Karhu, A. Impact of AIP and inhibitory G protein alpha 2 proteins on clinical features of sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2017, 176, 243–252.
- Puig-Domingo, M.; Gil, J.; Sampedro-Nunez, M.; Jorda, M.; Webb, S.M.; Serra, G.; Pons, L.; Salinas, I.; Blanco, A.; Marques-Pamies, M.; et al. Molecular profiling for acromegaly treatment: A validation study. Endocr. Relat. Cancer 2020, 27, 375–389.
- Tirosh, A.; Jin, D.X.; De Marco, L.; Laitman, Y.; Friedman, E. Activating genomic alterations in the Gs alpha gene (GNAS) in 274 694 tumors. Genes Chromosomes Cancer 2020, 59, 503–516.
- Jung, H.; Kim, K.; Kim, D.; Moon, J.H.; Kim, E.H.; Kim, S.H.; Ku, C.R.; Lee, E.J. Associations of GNAS mutations with surgical outcomes in patients with growth hormone-secreting pituitary adenoma. Endocrinol. Metab. 2021, 36, 342–350.
- Romanet, P.; Galluso, J.; Kamenicky, P.; Hage, M.; Theodoropoulou, M.; Roche, C.; Graillon, T.; Etchevers, H.C.; De Murat, D.; Mougel, G.; et al. Somatotroph tumors and the epigenetic status of the GNAS locus. Int. J. Mol. Sci. 2021, 22, 7570.
- Yamato, A.; Nagano, H.; Gao, Y.; Matsuda, T.; Hashimoto, N.; Nakayama, A.; Yamagata, K.; Yokoyama, M.; Gong, Y.; Shi, X.; et al. Proteogenomic landscape and clinical characterization of GH-producing pituitary adenomas/somatotroph pituitary neuroendocrine tumors. Commun. Biol. 2022, 5, 1304.
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424.
- Turan, S.; Bastepe, M. GNAS Spectrum of Disorders. Curr. Osteoporos. Rep. 2015, 13, 146–158.
- Ritterhouse, L.L.; Vivero, M.; Mino-Kenudson, M.; Sholl, L.M.; Iafrate, A.J.; Nardi, V.; Dong, F. GNAS mutations in primary mucinous and non-mucinous lung adenocarcinomas. Mod. Pathol. 2017, 30, 1720–1727.
- Faias, S.; Duarte, M.; Pereira, L.; Chaves, P.; Cravo, M.; Dias Pereira, A.; Albuquerque, C. Methylation changes at the GNAS imprinted locus in pancreatic cystic neoplasms are important for the diagnosis of malignant cysts. World J. Gastrointest. Oncol. 2020, 12, 1056–1064.
- McCune, D.J. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am. J. Dis. Child 1936, 52, 743–744.
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695.
- Boyce, A.M.; Florenzano, P.; de Castro, L.F.; Collins, M.T. Fibrous dysplasia/McCune-Albright syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019.
- Lumbroso, S.; Paris, F.; Sultan, C. Activating Gsalpha mutations: Analysis of 113 patients with signs of McCune-Albright syndrome—A European Collaborative Study. J. Clin. Endocrinol. Metab. 2004, 89, 2107–2113.
- Idowu, B.D.; Al-Adnani, M.; O’Donnell, P.; Yu, L.; Odell, E.; Diss, T.; Gale, R.E.; Flanagan, A.M. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: The first report of a codon 227 mutation in bone. Histopathology 2007, 50, 691–704.
- Spencer, T.; Pan, K.S.; Collins, M.T.; Boyce, A.M. The clinical spectrum of McCune-Albright syndrome and its management. Horm. Res. Paediatr. 2019, 92, 347–356.
- Boyce, A.M.; Collins, M.T. Fibrous dysplasia/McCune-Albright syndrome: A rare, mosaic disease of Galpha s activation. Endocr. Rev. 2020, 41, 345–370.
- Spada, A.; Vallar, L.; Faglia, G. G-proteins and hormonal signalling in human pituitary tumors: Genetic mutations and functional alterations. Front. Neuroendocrinol. 1993, 14, 214–232.
- Weinstein, L.S. The stimulatory G protein alpha-subunit gene: Mutations and imprinting lead to complex phenotypes. J. Clin. Endocrinol. Metab. 2001, 86, 4622–4626.
- Adams, E.F.; Lei, T.; Buchfelder, M.; Petersen, B.; Fahlbusch, R. Biochemical characteristics of human pituitary somatotropinomas with and without gsp mutations: In vitro cell culture studies. J. Clin. Endocrinol. Metab. 1995, 80, 2077–2081.
- Rymuza, J.; Kober, P.; Rusetska, N.; Mossakowska, B.J.; Maksymowicz, M.; Nyc, A.; Baluszek, S.; Zielinski, G.; Kunicki, J.; Bujko, M. Transcriptomic classification of pituitary neuroendocrine tumors causing acromegaly. Cells 2022, 11, 3846.
- Spada, A.; Arosio, M.; Bochicchio, D.; Bazzoni, N.; Vallar, L.; Bassetti, M.; Faglia, G. Clinical, biochemical, and morphological correlates in patients bearing growth hormone-secreting pituitary-tumors with or without constitutively active adenylyl cyclase. J. Clin. Endocr. Metab. 1990, 71, 1421–1426.
- Larkin, S.; Reddy, R.; Karavitaki, N.; Cudlip, S.; Wass, J.; Ansorge, O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur. J. Endocrinol. 2013, 168, 491–499.
- Lasolle, H.; Elsensohn, M.H.; Wierinckx, A.; Alix, E.; Bonnefille, C.; Vasiljevic, A.; Cortet, C.; Decoudier, B.; Sturm, N.; Gaillard, S.; et al. Chromosomal instability in the prediction of pituitary neuroendocrine tumors prognosis. Acta Neuropathol. Commun. 2020, 8, 190.
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 2020, 37, 123–134.e125.
- Mantovani, G.; Bondioni, S.; Lania, A.G.; Corbetta, S.; de Sanctis, L.; Cappa, M.; Di Battista, E.; Chanson, P.; Beck-Peccoz, P.; Spada, A. Parental origin of Gsalpha mutations in the McCune-Albright syndrome and in isolated endocrine tumors. J. Clin. Endocrinol. Metab. 2004, 89, 3007–3009.
- Hayward, B.E.; Barlier, A.; Korbonits, M.; Grossman, A.B.; Jacquet, P.; Enjalbert, A.; Bonthron, D.T. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J. Clin. Investig. 2001, 107, R31–R36.
- Picard, C.; Silvy, M.; Gerard, C.; Buffat, C.; Lavaque, E.; Figarella-Branger, D.; Dufour, H.; Gabert, J.; Beckers, A.; Brue, T.; et al. Gs alpha overexpression and loss of Gs alpha imprinting in human somatotroph adenomas: Association with tumor size and response to pharmacologic treatment. Int. J. Cancer 2007, 121, 1245–1252.
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151.
- Schultz, K.A.P.; Stewart, D.R.; Kamihara, J.; Bauer, A.J.; Merideth, M.A.; Stratton, P.; Huryn, L.A.; Harris, A.K.; Doros, L.; Field, A.; et al. DICER1 tumor predisposition. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020.
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009, 325, 965.
- Slade, I.; Bacchelli, C.; Davies, H.; Murray, A.; Abbaszadeh, F.; Hanks, S.; Barfoot, R.; Burke, A.; Chisholm, J.; Hewitt, M.; et al. DICER1 syndrome: Clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J. Med. Genet. 2011, 48, 273–278.
- de Kock, L.; Wang, Y.C.; Revil, T.; Badescu, D.; Rivera, B.; Sabbaghian, N.; Wu, M.; Weber, E.; Sandoval, C.; Hopman, S.M.; et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J. Med. Genet. 2016, 53, 43–52.
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 2018, 24, 2251–2261.
- Klein, S.; Lee, H.; Ghahremani, S.; Kempert, P.; Ischander, M.; Teitell, M.A.; Nelson, S.F.; Martinez-Agosto, J.A. Expanding the phenotype of mutations in DICER1: Mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J. Med. Genet. 2014, 51, 294–302.
- Brenneman, M.; Field, A.; Yang, J.; Williams, G.; Doros, L.; Rossi, C.; Schultz, K.A.; Rosenberg, A.; Ivanovich, J.; Turner, J.; et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/DICER1 syndrome: A unique variant of the two-hit tumor suppression model. F1000Res 2015, 4, 214.
- MacRae, I.J.; Zhou, K.; Li, F.; Repic, A.; Brooks, A.N.; Cande, W.Z.; Adams, P.D.; Doudna, J.A. Structural basis for double-stranded RNA processing by Dicer. Science 2006, 311, 195–198.
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366.
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672.
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744.
- Svoboda, P. Key mechanistic principles and considerations concerning RNA interference. Front. Plant Sci. 2020, 11, 1237.
- de Boer, C.M.; Eini, R.; Gillis, A.M.; Stoop, H.; Looijenga, L.H.; White, S.J. DICER1 RNase IIIb domain mutations are infrequent in testicular germ cell tumours. BMC. Res. Notes 2012, 5, 569.
- Heravi-Moussavi, A.; Anglesio, M.S.; Cheng, S.W.; Senz, J.; Yang, W.; Prentice, L.; Fejes, A.P.; Chow, C.; Tone, A.; Kalloger, S.E.; et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N. Engl. J. Med. 2012, 366, 234–242.
- de Kock, L.; Plourde, F.; Carter, M.T.; Hamel, N.; Srivastava, A.; Meyn, M.S.; Arseneau, J.; Bouron-Dal, S.D.; Foulkes, W.D. Germ-line and somatic DICER1 mutations in a pleuropulmonary blastoma. Pediatr. Blood Cancer 2013, 60, 2091–2092.
- Wu, M.K.; Sabbaghian, N.; Xu, B.; Addidou-Kalucki, S.; Bernard, C.; Zou, D.; Reeve, A.E.; Eccles, M.R.; Cole, C.; Choong, C.S.; et al. Biallelic DICER1 mutations occur in Wilms tumours. J. Pathol. 2013, 230, 154–164.
- Tomiak, E.; de, K.L.; Grynspan, D.; Ramphal, R.; Foulkes, W.D. DICER1 mutations in an adolescent with cervical embryonal rhabdomyosarcoma (cERMS). Pediatr. Blood Cancer 2014, 61, 568–569.
- de Kock, L.; Sabbaghian, N.; Soglio, D.B.D.; Guillerman, R.P.; Park, B.K.; Chami, R.; Deal, C.L.; Priest, J.R.; Foulkes, W.D. Exploring the association between DICER1 mutations and differentiated thyroid carcinoma. J. Clin. Endocr. Metab. 2014, 99, E1072–E1077.
- de Kock, L.; Sabbaghian, N.; Druker, H.; Weber, E.; Hamel, N.; Miller, S.; Choong, C.S.; Gottardo, N.G.; Kees, U.R.; Rednam, S.P.; et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta. Neuropathol. 2014, 128, 583–595.
- de Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta. Neuropathol. 2014, 128, 111–122.
- Doros, L.A.; Rossi, C.T.; Yang, J.; Field, A.; Williams, G.M.; Messinger, Y.; Cajaiba, M.M.; Perlman, E.J.; Schultz, A.; Cathro, H.P.; et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod. Pathol. 2014, 27, 1267–1280.
- Murray, M.J.; Bailey, S.; Raby, K.L.; Saini, H.K.; de, K.L.; Burke, G.A.; Foulkes, W.D.; Enright, A.J.; Coleman, N.; Tischkowitz, M. Serum levels of mature microRNAs in DICER1-mutated pleuropulmonary blastoma. Oncogenesis 2014, 3, e87.
- Pugh, T.J.; Yu, W.; Yang, J.; Field, A.L.; Ambrogio, L.; Carter, S.L.; Cibulskis, K.; Giannikopoulos, P.; Kiezun, A.; Kim, J.; et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 2014, 33, 5295–5302.
- Anglesio, M.S.; Wang, Y.; Yang, W.; Senz, J.; Wan, A.; Heravi-Moussavi, A.; Salamanca, C.; Maines-Bandiera, S.; Huntsman, D.G.; Morin, G.B. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J. Pathol. 2013, 229, 400–409.
- Chong, A.S.; Han, H.; Albrecht, S.; Weon, Y.C.; Park, S.K.; Foulkes, W.D. DICER1 syndrome in a young adult with pituitary blastoma. Acta. Neuropathol. 2021, 142, 1071–1076.
- Liu, A.P.Y.; Kelsey, M.M.; Sabbaghian, N.; Park, S.H.; Deal, C.L.; Esbenshade, A.J.; Ploner, O.; Peet, A.; Traunecker, H.; Ahmed, Y.H.E.; et al. Clinical outcomes and complications of pituitary blastoma. J. Clin. Endocrinol. Metab. 2021, 106, 351–363.
- Liu, A.P.; Li, K.K.; Chow, C.; Chan, S.; Leung, A.W.; Shing, M.M.; To, K.F.; Chan, D.T.; Chan, G.C.; Ng, H.K. Expanding the clinical and molecular spectrum of pituitary blastoma. Acta. Neuropathol. 2022, 143, 415–417.
- Scheithauer, B.W.; Kovacs, K.; Horvath, E.; Kim, D.S.; Osamura, R.Y.; Ketterling, R.P.; Lloyd, R.V.; Kim, O.L. Pituitary blastoma. Acta. Neuropathol. 2008, 116, 657–666.
- Nadaf, J.; de Kock, L.; Chong, A.S.; Korbonits, M.; Thorner, P.; Benlimame, N.; Fu, L.; Peet, A.; Warner, J.; Ploner, O.; et al. Molecular characterization of DICER1-mutated pituitary blastoma. Acta. Neuropathol. 2021, 141, 929–944.
- Asada, K.; Canestrari, E.; Fu, X.; Li, Z.; Makowski, E.; Wu, Y.C.; Mito, J.K.; Kirsch, D.G.; Baraban, J.; Paroo, Z. Rescuing dicer defects via inhibition of an anti-dicing nuclease. Cell Rep. 2014, 9, 1471–1481.
- Asada, K.; Canestrari, E.; Paroo, Z. A druggable target for rescuing microRNA defects. Bioorg Med. Chem. Lett. 2016, 26, 4942–4946.
- Muller, M.; Fazi, F.; Ciaudo, C. Argonaute proteins: From structure to function in development and pathological cell fate determination. Front Cell Dev. Biol. 2019, 7, 360.
- Thunders, M.; Delahunt, B. Gene of the month: DICER1: Ruler and controller. J. Clin. Pathol. 2021, 74, 69–72.
- Meiklejohn, K.M.; Darbinyan, A.; Barbieri, A.L. A review of DICER1: Structure, function and contribution to disease. Diagnostic Histopathology 2022, 28, 329–336.
- Cretu, C.; Schmitzova, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Luhrmann, R.; Pena, V. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol. Cell 2016, 64, 307–319.
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615.
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3’ splice site selection through use of a different branch point. Cell Rep. 2015, 13, 1033–1045.
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105.
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707.
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524.
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark Res. 2020, 8, 38.
- Li, C.; Xie, W.; Rosenblum, J.S.; Zhou, J.; Guo, J.; Miao, Y.; Shen, Y.; Wang, H.; Gong, L.; Li, M.; et al. Somatic SF3B1 hotspot mutation in prolactinomas. Nat. Commun. 2020, 11, 2506.
- Simon, J.; Perez-Rivas, L.G.; Zhao, Y.; Chasseloup, F.; Lasolle, H.; Cortet, C.; Descotes, F.; Villa, C.; Baussart, B.; Burman, P.; et al. Prevalence and clinical correlations of SF3B1 variants in lactotroph tumours. Eur. J. Endocrinol. 2023, 189, 372–378.
- Cass, D.M.; Berglund, J.A. The SF3b155 N-terminal domain is a scaffold important for splicing. Biochemistry 2006, 45, 10092–10101.
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395.
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506.
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69.
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405.
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404.
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135.
- Kong, Y.; Krauthammer, M.; Halaban, R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res. 2014, 24, 332–334.
- Yang, H.M.; Hsiao, S.J.; Schaeffer, D.F.; Lai, C.; Remotti, H.E.; Horst, D.; Mansukhani, M.M.; Horst, B.A. Identification of recurrent mutational events in anorectal melanoma. Mod. Pathol. 2017, 30, 286–296.
- Guo, J.; Li, C.; Fang, Q.; Liu, Y.; Wang, D.; Chen, Y.; Xie, W.; Zhang, Y. The SF3B1(R625H) mutation promotes prolactinoma tumor progression through aberrant splicing of DLG1. J. Exp. Clin. Cancer Res. 2022, 41, 26.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
556
Revisions:
2 times
(View History)
Update Date:
14 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No