Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shuya Kasai | -- | 2531 | 2023-12-13 01:23:51 | | | |
| 2 | Lindsay Dong | Meta information modification | 2531 | 2023-12-18 01:54:48 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2531 | 2023-12-18 01:55:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kasai, S.; Kokubu, D.; Mizukami, H.; Itoh, K. Nrf2 in Obesity-Related Insulin Resistance and T2D Prevention. Encyclopedia. Available online: https://encyclopedia.pub/entry/52634 (accessed on 12 June 2026).
Kasai S, Kokubu D, Mizukami H, Itoh K. Nrf2 in Obesity-Related Insulin Resistance and T2D Prevention. Encyclopedia. Available at: https://encyclopedia.pub/entry/52634. Accessed June 12, 2026.
Kasai, Shuya, Daichi Kokubu, Hiroki Mizukami, Ken Itoh. "Nrf2 in Obesity-Related Insulin Resistance and T2D Prevention" Encyclopedia, https://encyclopedia.pub/entry/52634 (accessed June 12, 2026).
Kasai, S., Kokubu, D., Mizukami, H., & Itoh, K. (2023, December 13). Nrf2 in Obesity-Related Insulin Resistance and T2D Prevention. In Encyclopedia. https://encyclopedia.pub/entry/52634
Kasai, Shuya, et al. "Nrf2 in Obesity-Related Insulin Resistance and T2D Prevention." Encyclopedia. Web. 13 December, 2023.
Copy Citation
Reactive oxygen species (ROS) are produced mainly by mitochondrial respiration and function as signaling molecules in the physiological range. However, ROS production is also associated with the pathogenesis of various diseases, including insulin resistance (IR) and type 2 diabetes (T2D).
mitochondrial ROS
Nrf2
insulin resistance
diabetes
1. Introduction
OxPhos proteins in the mitochondria are a mosaic of nuclear- and mitochondria-encoded proteins except for complex II, and electron transfer is not perfectly shielded. Therefore, a small percentage of electrons that evade from an electron transfer chain (ETC) cause a one-electron reduction in O2 either in the mitochondrial matrix or intermembrane space, leading to the generation of superoxide anion radicals. Superoxides are further reduced by superoxide dismutases SOD2 and SOD1 localized in the matrix or the intermembrane space, respectively, leading to the generation of hydrogen peroxides (H2O2) [1]. Mitochondrial H2O2 is reduced into water by glutathione peroxidase-1 and -4 (GPX-1/-4) and peroxiredoxin-3 (PRDX3) [2]. The generation of mitochondrial reactive oxygen species (mtROS) increases when the ETC is reduced, either by the increased influx of electrons to the ETC, such as in oxygen reperfusion injuries, or by the slowdown/inhibition of electron transfer, such as in decreased demand for ATP. These mtROS oxidize cellular components and cause damage when produced in excess, such as in ischemia-reperfusion injuries [3].
NF-E2-related factor 2 (Nrf2) is a transcription factor that regulates gene expression to elicit a defense system against oxidative stress in multicellular animals that evolved after metazoans [4][5]. Nrf2 activation is regulated by Kelch-like ECH-associated protein 1 (Keap1), which functions as a sensor for electrophiles and oxidative stress. Nrf2 target genes take part in detoxification, glutathione (GSH) synthesis, redox homeostasis, and proteostasis to counteract oxidative stress, mainly in the cytoplasm. Dietary Nrf2 inducers (e.g., sulforaphane)-triggered Nrf2-mediated oxidative stress response induction could modulate or prevent such disease progression. In fact, the Nrf2 inducer is used as a therapeutic agent for neurodegeneration and kidney disease associated with oxidative stress and inflammation [6][7][8]. On the other hand, somatic mutations that cause Nrf2 activation were found in tumors such as non-small cell lung epithelial and esophagus cancers, and Nrf2 plays tumorigenic functions [9][10][11].
2. Mitochondrial Dysfunction at the Common Pathway in T2D Pathogenesis
2.1. T2D Etiology
T2D is often associated with obesity and old age, manifesting impaired glucose tolerance and increased fasting blood glucose levels. Obesity, IR, and hyperinsulinemia together precede T2D as a triad [12]. However, the members of this triad are interrelated, and determining the initial cause that ultimately leads to T2D is difficult. In fact, chronic hyperinsulinemia leads to IR [13][14], and genetic manipulation-induced IR causes hyperinsulinemia in mice. The concept that overflowed free fatty acids, as a result of overnutrition, would cause IR (usually called lipotoxicity) has been heavily debated [15]. However, the precise underlying mechanisms of IR are unclear.
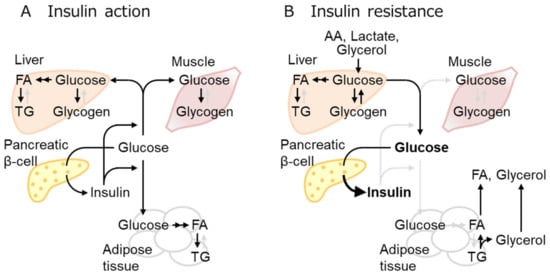
2.2. IR Mechanisms as the Early Event of Obese T2D
Insulin exerts overlapping but different roles in each organ. IR designates the defect of insulin to take action, although it generally refers to the inability of a certain amount of insulin to lower blood glucose levels mainly by glucose uptake in the muscles, liver, and fat as well as to suppress glucose production in the liver [12] (Figure 1).
Figure 1. Insulin effect and resistance in multiple tissues. (A) Increased blood glucose stimulates insulin secretion, thereby increasing glucose uptake mainly in the muscles, adipose tissue, and liver, where glucose is stored as glycogen or converted to fatty acid (FA), then triglyceride (TG). (B) Insulin resistance denotes the loss of the insulin effect, glucose uptake reduction in insulin-sensitive tissues, and impaired suppression of hepatic gluconeogenesis, leading to hyperglycemia. Three-carbon substrates, including amino acids (AA), lactate, and glycerol, are used for gluconeogenesis. Overnutrition increases fat mass and circulating FA over the adipose tissue capacities. Active and inactive metabolic pathways were indicated with black and pale gray arrows, respectively.
Obesity is often associated with adipose tissue-related inflammation. However, inflammation might not be the initial cause of IR, although it exacerbates IR, ultimately leading to T2D [12][16]. A growing body of evidence suggests that IR in T2D is not caused by the defect of proximal insulin signaling (such as the defect of AKT phosphorylation), but by the defect of distal insulin signaling located more closely to the final biological response (such as defects of GLUT4 trafficking to the plasma membrane) [12]. Insulin receptors and proximal signaling molecules (such as AKT) are available in excess, and the downregulation of these molecules, as reported in heterozygous knockouts (KO), does not affect the final signaling outcome.
2.3. BCAA Metabolism Reduction in WAT as the Earliest Response to Obesity-Related Hyperinsulinemia and IR
Overnutrition leads to adipose tissue mass expansion either by hypertrophy (adipocyte size increase) or hyperplasia (increase in adipocyte number by preadipocyte differentiation and adipogenesis) [17]. Hyperplasia is considered healthy obesity, displaying well-organized growth with vascularization and producing beneficial adipokines such as leptin and adiponectin. In contrast, hypertrophy (i.e., pathologic obesity) causes fibrosis, inflammation, and necrosis, and is associated with ectopic lipid deposition. Generally, adipogenesis is considered to be protective against obesity-associated T2D. Subjects with a PPARγ variant that reduces its adipogenesis capacity have an increased risk of developing T2D [18], and PPARγ ligands are actually utilized as drugs for T2D.
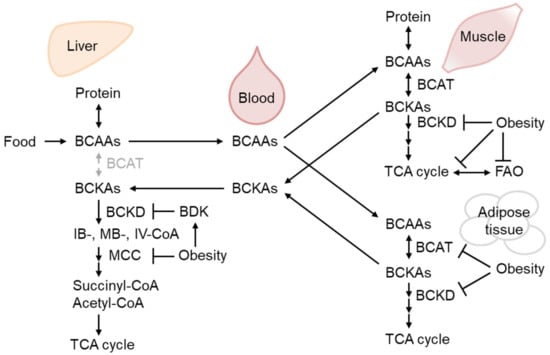
BCAAs are supplied by dietary ingredients, that are affected by gut microbiota metabolism, and by protein breakdown [19][20]. Importantly, adipogenesis utilizes a significant amount of BCAA as an energy source for preadipocyte differentiation [21], as do mature adipocytes [22]. After BCAAs are catabolized to corresponding ketoacids mainly in the muscle, branched-chain ketoacids are metabolized in the mitochondria by the branched-chain α-ketoacid dehydrogenase (BCKD) complex [23] (Figure 2).
Consistent with the above-mentioned reports, BCAA has emerged as the most reliable biomarker for obesity-associated IR and T2D [24], although a growing number of studies have demonstrated that BCAA supplementation positively affects metabolic health [25]. Pioneering metabolomic analysis demonstrated that BCAA or its metabolites are the most reliable markers of impaired fasting glucose after glucose in TwinsUK [24]. Plasma BCAA or their respective ketoacid metabolites are predictors of future T2D, with the latter being better markers.
Figure 2. The regulation of branched-chain amino acid (BCAA) catabolism in obesity. BCAAs from dietary sources and protein breakdown are mainly oxidized by branched-chain aminotransferase (BCAT) in the muscle to respective ketoacids (BCKAs), which could be further oxidized by BCKA dehydrogenase (BCKD) and metabolized eventually into TCA cycle substrates, succinyl-CoA, and acetyl-CoA. Obesity decreases BCAA-catabolizing enzyme activity in the liver and skeletal muscle as well as their expression in the adipose tissue, leading to BCAA and BCKA accumulation in the blood. BCKD activity is inhibited by phosphorylation by BCKD kinase (BDK), which is increased in the liver of obese mice [26][27].
3. The Role of mtROS in IR
3.1. mtROS as the Cause of IR
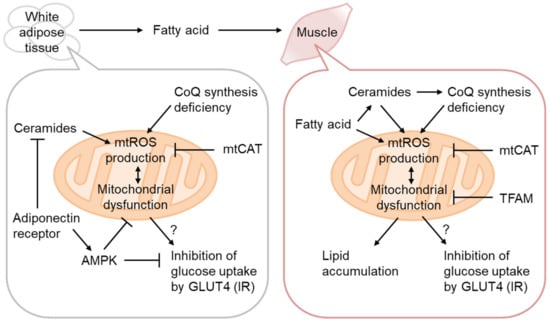
Mitochondrial dysfunction, oxidative stress, and lipid accumulation are often associated with IR in the skeletal muscle [28]. The causative role of mitochondrial dysfunction has thus been mostly examined in this tissue [29] (Figure 3). As decreased OxPhos and lipid accumulation without enhancement of systemic lipolysis and signs of inflammation have been observed in the muscles of prediabetic lean IR people of the offspring of T2D patients [30], mitochondrial dysfunction might be causative for IR.
Figure 3. Mitochondrial ROS production in IR. Coenzyme Q (CoQ) synthetic pathway downregulation in IR increases H2O2 production from Complex II, rescued by CoQ supplementation or mitochondria-targeting catalase (mtCAT) expression. Adipocyte hypertrophy attenuates adiponectin signaling and the ceramidase activity of the adiponectin receptor, resulting in ceramide accumulation and mitochondrial dysfunction. mtROS production in the skeletal muscle is increased by excess fatty acid oxidation, either directly or indirectly, via ceramide accumulation and CoQ depletion. H2O2 production by mitochondria-targeting paraquat (MitoParaquat) can mimic muscle IR, reversed by mtCAT or TFAM expression. Mitochondrial dysfunction, rather than AKT inhibition by ceramide, affects GLUT4 translocation to the plasma membrane and inhibits glucose uptake.
3.2. Ceramides as mtROS Sources in Insulin Resistance
Ceramides are generated via de novo biosynthesis, sphingomyelin hydrolysis, the catabolism of complex sphingolipids, etc. [31]. It has been known that ceramide synthesis is activated by various stresses, including inflammation and oxidative stress, and that it regulates apoptosis, etc., but recent reports have placed its central role in the regulation of metabolism. Actually, several studies have indicated that inhibition of ceramide synthesis prevents diet-induced IR. Ceramides may act as gauges for surplus FAs, as serine and palmitoyl-CoA are the starting substrates of de novo ceramide synthesis.
4. Role of Nrf2 in Prevention of Obesity-Related IR and T2D
4.1. The Regulation of Oxidative Stress Response by Nrf2
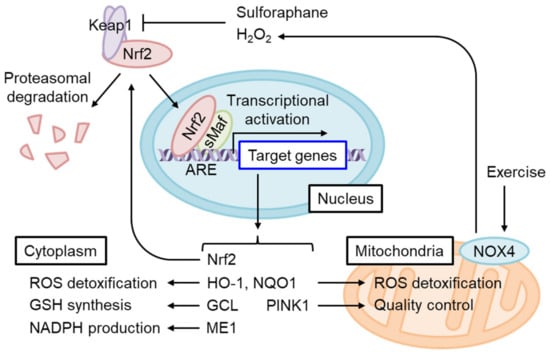
Nrf2 is a short-lived protein with a half-life of ~20 min under non-stressed conditions, submitted to Keap1-mediated ubiquitination and proteasomal degradation [4] (Figure 4). The oxidation of certain Keap1 cysteine residues by electrophiles or ROS induces Nrf2 accumulation and the dimerization of Nrf2 and small Maf (sMaf) in the nucleus. The Nrf2/sMaf heterodimer binds to the antioxidant response element (ARE) and induces the expression of various detoxification- and antioxidant synthesis-related genes (Figure 5).
Figure 4. Nrf2 activation restores cytoplasmic and mitochondrial oxidative stress in IR. Nrf2 constitutively undergoes Keap1-mediated ubiquitination and proteasomal degradation under physiological redox states. Keap1 is inhibited by the adduction or oxidation of specific cysteine residues by electrophiles and by oxidative stress. Sulforaphane or H2O2 produced by exercise-stimulated NOX4 stabilizes and activates Nrf2. A heterodimer of Nrf2 and small Maf (sMaf) binds the antioxidant response element (ARE) and induces target gene transcription. ARE is present in the Nrf2 gene enhancer and increases Nrf2 mRNA levels in an auto-regulatory loop. Nrf2 target genes include those encoding enzymes that detoxify cytoplasmic ROS as well as electrophiles and produce antioxidants, while other genes regulate mitochondrial redox homeostasis and quality control.
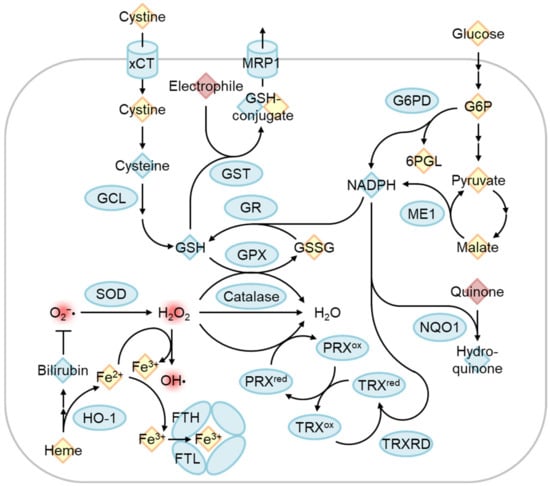
Figure 5. Regulation of oxidative stress response by Nrf2 target genes. The Nrf2-regulated gene product is indicated by the blue circle. Reactive oxygen species and electrophiles are indicated in red. Antioxidants and other chemicals were indicated as blue and yellow diamonds, respectively. Cystine/glutamate transporter (xCT) and glutamate-cysteine ligase (GCL) are involved in glutathione synthesis, and glutathione reductase recycles oxidized glutathione (GSSG) to reduced glutathione (GSH). GSH is used as an antioxidant, conjugation of electrophile by glutathione S-transferase (GST) to excrete via multidrug resistance-associated protein (MRP1), and reduction of H2O2 to H2O by glutathione peroxidase (GPX). H2O2 can be produced by superoxide (O2−•) dismutase (SOD) and is eliminated by the catalase and peroxiredoxin (PRX)/thioredoxin (TRX) systems. Free heme is degraded by heme oxygenase-1 (HO-1), and the degradation product bilirubin functions as an antioxidant. Released ferrous iron (Fe2+) can convert H2O2 into a hydroxyl radical (OH•) as known as the Fenton reaction. Fe2+ is oxidized to ferric iron (Fe3+) by the ferritin heavy chain (FTH) and stored inactively. Glucose-6-phosphate (G6P) dehydrogenase (G6PD) and malic enzyme 1 (ME1) produce NADPH, which is used for reactions by GR, TRX reductase (TRXRD), and NAD(P)H Quinone oxidoreductase 1 (NQO1). G6PD is a pentose phosphate pathway enzyme that produces 6-phosphogluconolactone (6PGL).
4.2. The Role of Nrf2 Activation in WAT and Obesity
Several studies have examined how Nrf2 could affect obesity or IR using systemic Nrf2 or Keap1 knockdown (KD) mice (i.e., constitutive Nrf2 activation model). Both mice are reportedly resistant to HFD-induced obesity, with some contradicting results in the case of the former [32]. As Nrf2 in adipocytes and preadipocytes contributes to lipid metabolism, differentiation, and adipogenesis conducted by C/EBPβ and RXRα, Nrf2 is expectably involved in adipogenesis in response to HFD [33][34]. Although Nrf2 is generally required for HFD-induced obesity, as mentioned, constitutive Nrf2 activation by Keap1 KD also inhibited HFD-induced obesity by decreasing PPARγ and C/EBPα [35]. As Nrf2 KO affects HFD-induced obesity, the role of HFD-induced IR is impossible to analyze using this model without negating the effect of obesity. However, tissue-specific Nrf2 KO in adipocytes, but not in hepatocytes, exacerbated HFD-induced IR [36].
4.3. The Role of Nrf2 in Pancreatic β-Cells
Pancreatic β-cells are known to be susceptible to oxidative stress due to the low-level expression of antioxidant enzymes and high ROS production coupled with glucose metabolism and insulin biosynthesis [37][38]. Baumel-Alterzon et al. showed that Nrf2 is activated by high glucose in ex vivo islet β-cells and also in mice fed HFD for 1 week [77]. β-cell-specific Nrf2 KO mice fed HFD exhibited increased oxidative stress, decreased β-cell mass, and worsened glucose tolerance. In contrast, either β-cell-specific Keap1 KO or administration of Nrf2 activator conferred resistance to β-cell dysfunction by HFD, implicating that Nrf2 pathway contributes to the protection and therapeutic prevention of β-cells from oxidative stress [77].
4.4. The Role of Nrf2 in Mitochondrial Function
Nrf2 target genes mainly act in the protection of cytoplasmic oxidative stress, although certain target genes such as HO-1, NQO1, and Pink1 might directly modify oxidative stress in the mitochondria [39] (Figure 4). On the other hand, excess mtROS, such as H2O2, might diffuse and cause oxidative stress in the cytoplasm. On this occasion, Nrf2 may protect against oxidative stress via the generation of GSH and upregulation of ROS detoxifying enzymes such as GPXs. However, excess ROS in some cases reduce Nrf2 translation, and therefore the Nrf2-mediated stress response is limited [40].
4.5. Insulin Resistance Alleviation upon Nrf2 Activation
Nrf2 activation by Keap1 knockdown improves glucose intolerance by enhancing glycogen branching [41]. Indeed, SFN improves IR in HFD-fed mice [42][43]. Xirouchaki et al. demonstrated that NADPH oxidase 4 (NOX4) is induced by moderate or intense acute stress or exercise training in mice and humans that precedes NOX2 induction when it occurs [44]. They demonstrated that skeletal muscle-specific NOX4 decreases Nrf2, mitochondrial biogenesis-related genes, including PPARα, and mitochondrial biogenesis itself in in vitro cultured myotubes.
4.6. The Effect of Aging on T2D Etiology
Aging accelerates multiple age-related diseases, including T2D. Aging reportedly downregulates various cytoprotective factors, including the above-mentioned coenzyme Q [45] and Nrf2 [46]. Importantly, aging deteriorates mitochondrial function, as described in Harman’s free radical theory of aging. Among others, aging particularly deteriorates Complex I or IV activities. As Complex I mostly consists of nuclear- and mitochondrial-encoded subunits, reasonably efficient Complex I function is susceptible to aging.
5. Conclusions
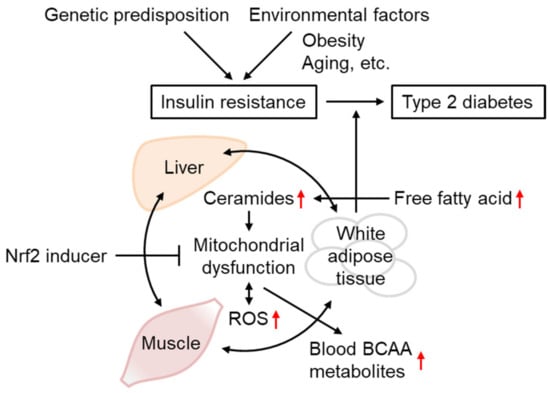
Various results obtained in IR models support the importance of mtROS overproduction in IR and WAT metabolic abnormalities in disease progression toward other tissues and T2D (Figure 6). Although the functional role of Nrf2 in IR remains controversial based on studies of Nrf2-deficient animals, Nrf2 activation by SFN and other inducers is concordantly beneficial for IR in insulin-sensitive tissues, including WAT. As the accumulation of BCAA and its metabolites in the blood reflects mitochondrial metabolism in the muscles and WAT in the case of obesity, the administration of Nrf2 inducers might provide a potential preventive intervention strategy against IR and T2D.
Figure 6. Insulin resistance progression and its prevention by Nrf2 inducers. The occurrence and progression of insulin resistance are affected both by genetic predisposition and environmental factors, including obesity and aging. IR is triggered by mitochondrial dysfunction and ROS overproduction, associated with ceramide accumulation. As IR in one tissue could propagate to other tissues, IR in the WAT and unhealthy obesity increase circulating free fatty acid levels, leading to depositions in the liver and muscles or conversion into ceramides, worsening the conditions that lead to type-2 diabetes. Defects in mitochondrial BCAA catabolism result in BCAA metabolite accumulation in the blood. Nrf2 inducer administration could improve IR through gene expression involved in antioxidant systems, mitochondrial function, and lipid metabolism.
References
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77.
- Ayer, A.; Fazakerley, D.J.; James, D.E.; Stocker, R. The role of mitochondrial reactive oxygen species in insulin resistance. Free Radic. Biol. Med. 2022, 179, 339–362.
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428.
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox. Signal. 2010, 13, 1665–1678.
- Gacesa, R.; Dunlap, W.C.; Barlow, D.J.; Laskowski, R.A.; Long, P.F. Rising levels of atmospheric oxygen and evolution of Nrf2. Sci. Rep. 2016, 6, 27740.
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575.
- Abdul-Muneer, P.M. Nrf2 as a Potential Therapeutic Target for Traumatic Brain Injury. J. Integr. Neurosci. 2023, 22, 81.
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative Stress and NRF2/KEAP1/ARE Pathway in Diabetic Kidney Disease (DKD): New Perspectives. Biomolecules 2022, 12, 1227.
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663.
- Ghareghomi, S.; Habibi-Rezaei, M.; Arese, M.; Saso, L.; Moosavi-Movahedi, A.A. Nrf2 Modulation in Breast Cancer. Biomedicines 2022, 10, 2668.
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Cellular Modulators of the NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Front. Biosci. 2023, 28, 143.
- James, D.E.; Stockli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771.
- Gray, S.L.; Donald, C.; Jetha, A.; Covey, S.D.; Kieffer, T.J. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology 2010, 151, 4178–4186.
- Rajan, S.; Shankar, K.; Beg, M.; Varshney, S.; Gupta, A.; Srivastava, A.; Kumar, D.; Mishra, R.K.; Hussain, Z.; Gayen, J.R.; et al. Chronic hyperinsulinemia reduces insulin sensitivity and metabolic functions of brown adipocyte. J. Endocrinol. 2016, 230, 275–290.
- Perseghin, G.; Scifo, P.; De Cobelli, F.; Pagliato, E.; Battezzati, A.; Arcelloni, C.; Vanzulli, A.; Testolin, G.; Pozza, G.; Del Maschio, A.; et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606.
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483.
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258.
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Rosen, E.D.; Altshuler, D. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132.
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583.
- Gojda, J.; Cahova, M. Gut Microbiota as the Link between Elevated BCAA Serum Levels and Insulin Resistance. Biomolecules 2021, 11, 1414.
- Zaganjor, E.; Yoon, H.; Spinelli, J.B.; Nunn, E.R.; Laurent, G.; Keskinidis, P.; Sivaloganathan, S.; Joshi, S.; Notarangelo, G.; Mulei, S.; et al. SIRT4 is an early regulator of branched-chain amino acid catabolism that promotes adipogenesis. Cell Rep. 2021, 36, 109345.
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21.
- Holecek, M. The role of skeletal muscle in the pathogenesis of altered concentrations of branched-chain amino acids (valine, leucine, and isoleucine) in liver cirrhosis, diabetes, and other diseases. Physiol. Res. 2021, 70, 293–305.
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.; Kastenmuller, G.; Shin, S.Y.; Petersen, A.K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276.
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736.
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1552–E1563.
- Lian, K.; Du, C.; Liu, Y.; Zhu, D.; Yan, W.; Zhang, H.; Hong, Z.; Liu, P.; Zhang, L.; Pei, H.; et al. Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes 2015, 64, 49–59.
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948.
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330.
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671.
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230.
- Li, S.; Eguchi, N.; Lau, H.; Ichii, H. The Role of the Nrf2 Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6973.
- Wang, Z.; Zuo, Z.; Li, L.; Ren, S.; Gao, T.; Fu, J.; Hou, Y.; Chen, Y.; Pi, J. Nrf2 in adipocytes. Arch. Pharm. Res. 2020, 43, 350–360.
- Annie-Mathew, A.S.; Prem-Santhosh, S.; Jayasuriya, R.; Ganesh, G.; Ramkumar, K.M.; Sarada, D.V.L. The pivotal role of Nrf2 activators in adipocyte biology. Pharmacol. Res. 2021, 173, 105853.
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218.
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195.
- Elksnis, A.; Martinell, M.; Eriksson, O.; Espes, D. Heterogeneity of Metabolic Defects in Type 2 Diabetes and Its Relation to Reactive Oxygen Species and Alterations in Beta-Cell Mass. Front. Physiol. 2019, 10, 107.
- Baumel-Alterzon, S.; Katz, L.S.; Brill, G.; Garcia-Ocana, A.; Scott, D.K. Nrf2: The Master and Captain of Beta Cell Fate. Trends Endocrinol. Metab. 2021, 32, 7–19.
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97.
- Pensabene, K.M.; LaMorte, J.; Allender, A.E.; Wehr, J.; Kaur, P.; Savage, M.; Eggler, A.L. Acute Oxidative Stress Can Paradoxically Suppress Human NRF2 Protein Synthesis by Inhibiting Global Protein Translation. Antioxidants 2023, 12, 1735.
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell Biol. 2016, 36, 1655–1672.
- Zhang, Y.; Wu, Q.; Liu, J.; Zhang, Z.; Ma, X.; Zhang, Y.; Zhu, J.; Thring, R.W.; Wu, M.; Gao, Y.; et al. Sulforaphane alleviates high fat diet-induced insulin resistance via AMPK/Nrf2/GPx4 axis. Biomed. Pharmacother. 2022, 152, 113273.
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin Ameliorates Obesity and Insulin Resistance through Adipose Tissue Browning and Reduction of Metabolic Endotoxemia in Mice. Diabetes 2017, 66, 1222–1236.
- Xirouchaki, C.E.; Jia, Y.; McGrath, M.J.; Greatorex, S.; Tran, M.; Merry, T.L.; Hong, D.; Eramo, M.J.; Broome, S.C.; Woodhead, J.S.T.; et al. Skeletal muscle NOX4 is required for adaptive responses that prevent insulin resistance. Sci. Adv. 2021, 7, eabl4988.
- Díaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The Paradox of Coenzyme Q(10) in Aging. Nutrients 2019, 11, 2221.
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
473
Revisions:
3 times
(View History)
Update Date:
18 Dec 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No