Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eleonóra Spekker | -- | 2847 | 2023-12-05 12:35:17 | | | |
| 2 | Peter Tang | + 2 word(s) | 2849 | 2023-12-06 01:55:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Spekker, E.; Nagy-Grócz, G.; Vécsei, L. Ion Channel Disturbances in Migraine Headache. Encyclopedia. Available online: https://encyclopedia.pub/entry/52380 (accessed on 10 August 2026).
Spekker E, Nagy-Grócz G, Vécsei L. Ion Channel Disturbances in Migraine Headache. Encyclopedia. Available at: https://encyclopedia.pub/entry/52380. Accessed August 10, 2026.
Spekker, Eleonóra, Gábor Nagy-Grócz, László Vécsei. "Ion Channel Disturbances in Migraine Headache" Encyclopedia, https://encyclopedia.pub/entry/52380 (accessed August 10, 2026).
Spekker, E., Nagy-Grócz, G., & Vécsei, L. (2023, December 05). Ion Channel Disturbances in Migraine Headache. In Encyclopedia. https://encyclopedia.pub/entry/52380
Spekker, Eleonóra, et al. "Ion Channel Disturbances in Migraine Headache." Encyclopedia. Web. 05 December, 2023.
Copy Citation
Migraine is a primary headache disorder, which is an enormous burden to the healthcare system. While some aspects of the pathomechanism of migraines remain unknown, the most accepted theory is that activation and sensitization of the trigeminovascular system are essential during migraine attacks. It has been suggested that ion channels may be important participants in the pathogenesis of migraine. Numerous ion channels are expressed in the peripheral and central nervous systems, including the trigeminovascular system, affecting neuron excitability, synaptic energy homeostasis, inflammatory signaling, and pain sensation. Dysfunction of ion channels could result in neuronal excitability and peripheral or central sensitization.
migraine
ion channels
potassium channels
ASICs
purinerg system
kynurenic system
glutamate
trigeminovascular system
1. Introduction
Migraine is a primary headache disorder affecting more than 15% of the world’s adult population during their most productive years, resulting in a global health and economic burden of billions of dollars. The clinical manifestation of migraine involves recurrent attacks accompanied by various associated symptoms [1] Despite intensive research efforts, the underlying processes of the disease are still the subject of ongoing investigations.
Altered ion channel function is implicated in several neurological disorders, and as such, the importance of ion channels in the pathogenesis of migraine has received significant attention in recent decades. Ion channels, especially potassium, sodium, and calcium channels expressed in various regions of the brain, play a role in neuronal signal transmission and the regulation of vascular tone. Dysregulation of these channels may contribute to the processes that trigger migraine attacks. For instance, disruptions in potassium channels can contribute to heightened neuronal excitability [2]. The sudden and synchronized activity of nerve cells, induced by abnormalities in potassium channels, has the potential to lead to headaches and other migraine symptoms [3]. Sodium channels participate in the formation of action potentials [4], while calcium channels regulate the release of neurotransmitters [5]; thus, issues in the regulation of these channels may increase neuronal activity and vascular changes [6].
2. Migraine Pathogenesis and the Impact of the Ion Channels
Migraine is one of the most common neurological disorders, characterized by a moderate or severe headache felt as a throbbing pain on one side of the head. Nausea is common for many migraine patients, with some experiencing vomiting during these episodes. Individuals undergoing a migraine headache tend to become more sensitive to light, sound, and odors. Additionally, some may encounter dizziness or problems with balance during a migraine attack. Furthermore, intensive exercise and physical exertion can exacerbate the severity of headaches [1]. It affects more than one billion individuals across the world, with a 3:1 prevalence in women [1]. According to the Global Burden of Disease Study 2016, migraine ranks as the second most prevalent cause of disability [7].
Although certain aspects of the pathomechanism of migraine are not yet known, the most accepted theory is that activation and sensitization of the trigeminovascular system (TVS) are essential during migraine attacks [8]. This leads to the liberation of neurotransmitters like calcitonin gene-related peptide (CGRP), substance P (SP), pituitary adenylate cyclase-activating polypeptide (PACAP), and neurokinin A (NKA) from primary sensory neurons. These neurotransmitters trigger mast cell degranulation and plasma extravasation [9][10]. Simultaneously, second-order neurons become activated in the caudal trigeminal nucleus (TNC), and their axons ascend to the thalamus, projecting nociceptive information to the primary somatosensory cortex [11].
Some migraineurs experience an aura during migraine attacks, which is a manifestation of temporary visual and somatosensory disturbances caused by cortical spreading depression (CSD)—a slowly spreading wave of depolarization of neurons and glia in the cortex. The aura can encompass not only visual and sensory symptoms but also motor and brainstem symptoms, such as muscle weakness, speech problems, dizziness, or balance disturbances [12][13]. It has been suggested that high extracellular levels of glutamate and K+ may be responsible for the propagation of CSD [14]. CSD can activate sensory neurons in the trigeminal ganglia (TG), suggesting the central (CNS) and peripheral nervous system (PNS) have a role in migraine [15]. Following CSD, molecules such as ATP, glutamate, K+, H+, arachidonic acid (AA), and nitric oxide (NO) are released locally and are thought to diffuse to and activate meningeal nociceptive neurons [16][17][18]; this leads to a localized rise in neuroactive inflammatory mediators and the sensitization of brainstem regions relevant to pain [19][20].
The trigeminocervical complex (TCC) makes direct connections with the periaqueductal gray (PAG) and areas of the rostral ventromedial medulla (RVM), including the nucleus raphe magnus (NRM), nucleus raphe dorsalis (DR), and locus coeruleus (LC) [10][21]. These nuclei affect TNC activity, and they have a role in pain transmission [22][23]. In addition, the TCC also sends direct projections to higher structures, such as the hypothalamus and thalamus, and from there, the incoming signal projects to the cortex [23]. The hypothalamus establishes direct connections with various structures implicated in pain processing, including the nucleus tractus solitarius, rostral ventromedial medulla, PAG, and NRM [24]. Moreover, dural nociceptive stimulation activates several hypothalamic nuclei [25]. As a result of a dural stimulus, the neurons of the TVS become mechanically hypersensitive; the reason for this may be that the migraine headache is throbbing in nature and intensifies when coughing or bending [10][26] (Figure 1).
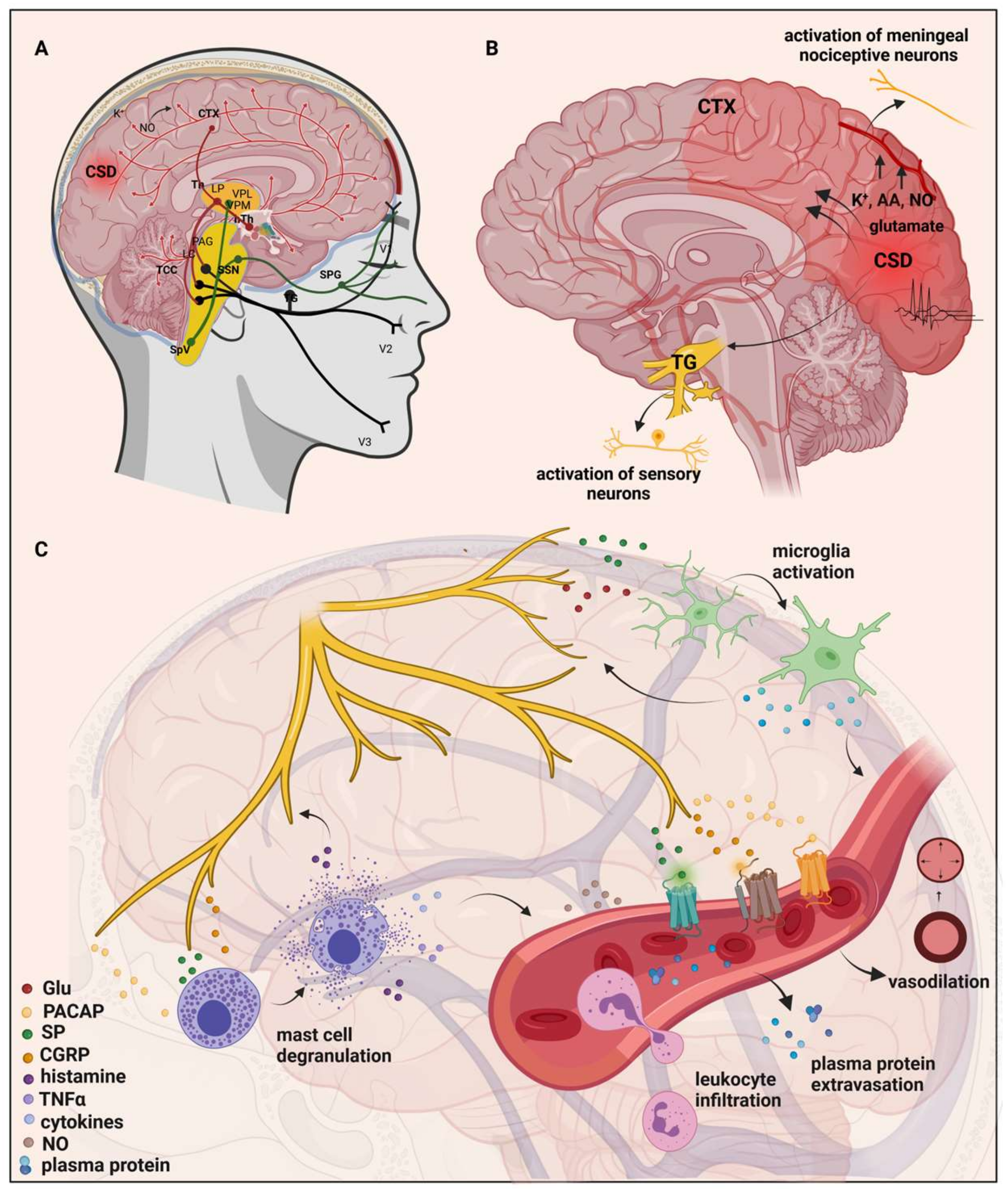
Figure 1. Mechanisms and structures involved in the pathogenesis of migraine. (A) Many brain regions are affected during migraine, such as the dorsolateral pons and dorsal midbrain: NRM, DR, LC, and PAG. These nuclei may influence the activity of the TNC and are involved in pain transmission. Moreover, apart from the TVS, they have a two-way connection with the thalamus and hypothalamus. (B) Initiation and propagation of CSD are determined by massive increases in extracellular K+, NO, and glutamate concentrations. CSD can activate the sensory neurons in trigeminal ganglia, and molecules such as ATP, glutamate, K+, H+, AA, and NO are released locally and are thought to diffuse to and activate meningeal nociceptive neurons. As a result, there is a local increase in neuroactive inflammatory mediators and sensitization of brainstem regions relevant to pain. (C) Stimulation of the trigeminal nerve causes the release of neuropeptides, leading to neurogenic inflammation. It has four main features: vasodilation and increased vascular permeability, leukocyte infiltration, activation of glial cells, and mast cell degranulation which results in increased production of inflammatory mediators such as cytokines and chemokines. AA, arachidonic acid; CTX, cortex; NO, nitric oxide; CSD, cortical spreading depression; Th, thalamus; hTh, hypothalamus; LP, lateral posterior nucleus; VPM, ventral posteromedial nucleus; VPL, ventral posterolateral nucleus; PAG, periaqueductal grey matter; LC, locus coeruleus; TCC, trigeminocervical complex; SSN, superior salivatory nucleus; SpV, spinal trigeminal nucleus caudalis; TG, trigeminal ganglion; SPG, sphenopalatine ganglion; V1, ophthalmic nerve; V2, maxillary nerve; V3, mandibular nerve; Glu, glutamate; CGRP, calcitonin gene-related peptide; SP, substance P; PACAP, pituitary adenylate cyclase-activating polypeptide; TNFα, tumor necrosis factor alpha; NRM, nucleus raphe magnus; DR, nucleus raphe dorsalis.
In recent decades, the importance of ion channels in the pathogenesis of migraine has received considerable attention, as an altered ion channel function can be observed in many neurological diseases [27]. Dysfunction or abnormal regulation of ion channels can lead to disruption of excitatory–inhibitory balance, neuronal excitability, and peripheral or central sensitization [28]. Genetic studies have identified several ion channel genes, including CACNA1A, ATP1A2, and SCN1A, which encode ion channels and transport proteins, as possible causes or contributors to familial hemiplegic migraine (FHM) [29][30][31][32]. Their function as ion channels and their involvement in ion transport, along with functional experiments in diverse cell and animal models, have played a part in revealing that their malfunction might play a role in cortical hyperexcitability and migraine.
3. Ion Channels in Migraine: Unraveling Pathogenesis and Therapeutic Implications
Ion channels are large membrane-spanning proteins that enable the selective transport of ions, such as potassium, calcium, and sodium. They mediate cell excitability and are essential for proper signaling and cell function [28]. Two types of ion channels can be distinguished, which open in response to changes in the membrane potential; these are voltage-gated ion channels (VGICs) and those that are opened by the binding of a ligand, such as a hormone or a neurotransmitter; these are ligand-dependent ion channels (LGICs) [33].
The activity of VGICs is modulated by the membrane potential of the cells. When the channels are open, they allow the movement of ions along an electrochemical gradient across cell membranes [34]. VGICs are selectively permeable to the main physiological ions (Na+, K+, Ca2+, and Cl−) and play an essential role in the generation and promotion of information in the form of action potentials in the CNS and PNS, as well as in the cardiovascular system [4].
LGICs mediate fast synaptic transmission in the nervous system and the somatic neuromuscular junction. The binding of a neurotransmitter to an orthosteric site causes a conformational change in the LGICs, and the channels are opened or gated. Gating can be modulated by binding endogenous or exogenous modulators to allosteric sites [35].
The VGICs allow the permeation of only one type of ion, while the LGICs are less selective and allow the permeation of two or more types of ions through the channel pore [33] (Figure 2).
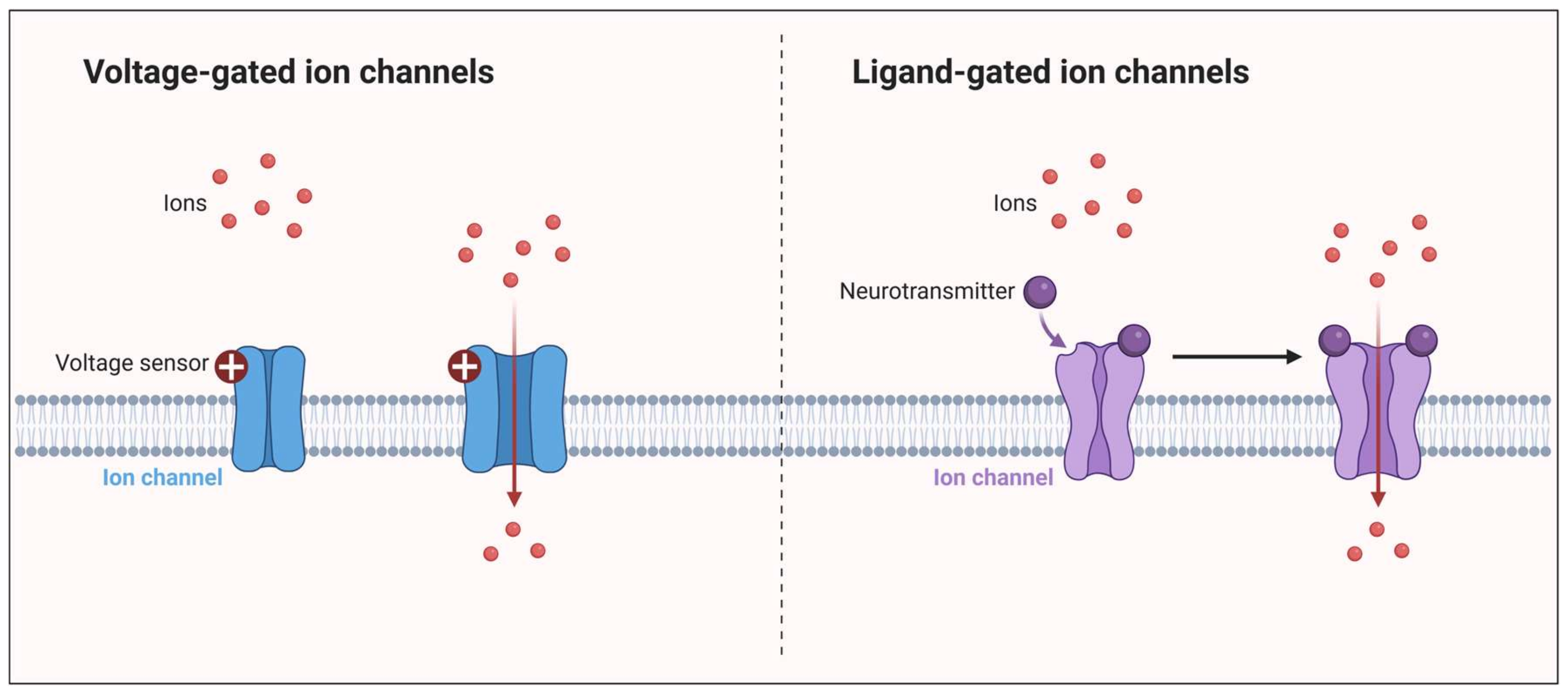
Figure 2. Ion channels: VGICs and LGICs. VGICs have a voltage-sensing domain. After a change in membrane potential, the channel opens and lets the ions flow through. LGICs have a ligand-binding domain. After the binding of the neurotransmitter, a conformational change occurs in the channel, and the free flow of ions occurs through it.
4. The Interplay of Glutamate and the Kynurenine Pathway in Migraine
4.1. Glutamate and Its Receptors
The glutamatergic system is a crucial neurotransmitter system in the brain that involves the neurotransmitter glutamate. Glutamate is the most abundant excitatory neurotransmitter in the CNS and plays a fundamental role in various brain functions, including learning, memory, cognition, neural plasticity, and pain transmission. The receptors of the glutamatergic system are divided into ionotropic and metabotropic receptors. Ionotropic receptors directly mediate the flow of ions across the cell membrane when glutamate binds to them. The three main types of ionotropic glutamate receptors are NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors. Activating these receptors is essential for processes like fast synaptic transmission and synaptic plasticity. Metabotropic receptors are coupled to intracellular signaling pathways through G-proteins and do not directly mediate ion flow. Instead, they modulate neuronal excitability and can have longer-lasting effects on synaptic transmission and plasticity.
Dysregulation of the glutamatergic system has been implicated in various neurological and neuropsychiatric disorders. For example, excessive glutamate release and subsequent overactivation of glutamate receptors can lead to excitotoxicity, a process associated with neurodegenerative diseases like Alzheimer’s disease and Parkinson’s disease, as reviewed by Szalárdy and his colleagues in 2012 [36]. Additionally, abnormalities in the glutamate receptor function have been linked to conditions like schizophrenia, mood disorders, and migraine disorders [37][38][39] as well. Elevated levels of glutamate have been found in the blood and cerebrospinal fluid in patients with migraine [40]. Glutamate excitotoxicity is associated with the hyperexcitability of NMDA receptors [41], which means that high glutamate stimulation causes an excessive amount of calcium ions to enter cells [42]. These processes have a crucial role in damaging DNA and different cell structures, yielding neuronal cell death. These receptors, principally the NMDA receptors, have an essential role in the pathomechanism of migraine.
The exact function of metabotropic receptors of glutamate in relation to migraines is not well understood. However, it is generally accepted that these receptors categorized under group I primarily contribute to the perception of pain [43]. This is because they are situated postsynaptically and, when activated, they heighten the brain’s responsiveness to stimuli. Conversely, metabotropic glutamate receptors in groups II and III are positioned presynaptically, and they work to decrease the release of glutamate, resulting in a mainly pain-relieving effect.
4.2. The Kynurenine Pathway
The kynurenine system is a biochemical pathway that involves the metabolism of the amino acid tryptophan. Tryptophan is an essential amino acid, which means that it must be obtained from the diet since the human body cannot synthesize it on its own. The kynurenine pathway is a major route through which tryptophan is metabolized, leading to the production of various metabolites with diverse physiological and immunological functions. The kynurenine pathway starts with the conversion of tryptophan to N-formyl-L-kynurenine by the enzyme indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO), depending on the tissue and the context. N-formyl-L-kynurenine is then further metabolized into L-kynurenine (L-KYN) by formamidase. L-KYN can also be metabolized to kynurenic acid (KYNA) by kynurenine aminotransferases, to anthranilic acid (ANA) by L-kynurenine hydrolase (KYNU), or to 3-hydroxy-L-kynurenine (3-HK) by kynurenine 3-monooxygenase (KMO) as well. ANA and 3-HK are then further degraded to 3-hydroxyanthranilic acid (3-HA), which metabolizes to quinolinic acid (QUIN). 3-HK can be metabolized to xanthurenic acid as well. As the last step of the kynurenine pathway, QUIN is converted to nicotinamide adenine dinucleotide (NAD+).
Kynurenines, particularly KYNA, have been identified as endogenous glutamate receptor antagonists. In line with this, KYNA acts as an opposing agent at the strychnine-insensitive glycine-binding site of NMDARs at lower concentrations [44]. Conversely, at higher doses, it also functions by obstructing the glutamate-binding site of NMDA receptors [45]. Furthermore, KYNA elicits mild opposing responses in relation to kainate- and AMPA-sensitive glutamate receptors [43]. Its influence on AMPA receptor-mediated activity is subject to concentration, demonstrating enhancement at lower levels (ranging from nanomolar to micromolar) and inhibition at elevated levels (ranging from micromolar to millimolar) [46]. This Janus-face effect has also been proven by electrophysiological investigations on the hippocampus of young rats, so KYNA actually enhances field excitatory postsynaptic potentials [47].
4.3. The Role of Kynurenine Pathway in Migraine Pathomechanism Connected to Glutamate Receptors
Several animal investigations suggest that kynurenines, as well as their analogs and halogenated derivatives, hold promise as potential therapeutic agents for treating migraines. Due to KYNA’s limited ability to traverse the blood–brain barrier, its analogs and derivatives are under experimental evaluation. Specifically, 4,6-dichlorokynurenine and 4-chlorokynurenine halogenated derivatives are converted into KYNA derivatives (7-chlorokynurenic acid and 5,7-dichlorokynurenic acid), which exhibit heightened affinity for the glycine-binding site of NMDA receptors [48][49].
In animal studies, the administration of L-KYN and probenecid (an inhibitor of KYNA secretion from the CNS) or KYNA analogs (N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride (KA1) and N-(2-N-pyrrolidinylethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride (KA2) effectively inhibited NTG-induced morphological and behavioral changes, likely by targeting NMDA receptors [50][51][52]. This model revealed decreased expression of kynurenine aminotransferase II (KATII), the primary enzyme in KYNA production, upon NTG administration [53]. Recent research has indicated that NTG influences the expression of other kynurenine pathway enzymes (TDO, IDO, KYNU, and KMO), implying an impact on the kynurenine pathway [54].
Another animal model involving trigeminal activation and sensitization includes the application of Complete Freund’s Adjuvant (CFA) to the dural surface, inducing inflammation. In this setup, KA1 was observed to alleviate CFA-induced inflammation [55]. Moreover, the researchers' group has shown that inflammatory soup could induce sterile neurogenic inflammation in the dura mater, leading to an expansion in the region affected by CGRP and transient receptor potential vanilloid 1 (TRPV1) reactive nerve fibers. Furthermore, there was an increase in the count of neuronal nitric oxide synthase (nNOS)-positive cells in the TNC. Prior applications of KYNA exhibited the capacity to regulate the alterations triggered by the inflammatory soup [56]. In the CFA model, the researchers' group also demonstrated that there was a sustained elevation in the levels of glutamate, KYNA, and L-KYN within the TNC 24 h following CFA treatment. Additionally, in the somatosensory cortex, the researchers observed significant increases in the concentrations of KYNA and serotonin, which strengthens the idea that inflammation can influence the elements of the glutamate and kynurenine system [57].
The orofacial formalin test, a model for simulating trigeminal activation and sensitization, demonstrated that probenecid reduced nociceptive behavior in rats by potentially increasing KYNA levels [58]. Recent studies using KA1 and KA2 abolished formalin-induced behavioral and morphological changes, elevating KYNA levels [59]. Additionally, in the combined NTG and formalin model, KA1 inhibited behavioral and morphological alterations [60]. In a trigeminal activation electrical stimulation model, reduced KAT immunoreactivity was observed in the rat’s dura mater [61].
In a CSD model, KA1 and KA2 inhibited CSD wave propagation, likely by targeting glutamate receptors, which play a pivotal role in CSD generation [62], potentially connecting migraine and CSD.
Stimulation of the trigeminal ganglion with electrical impulses led to notable elevations in levels of pituitary adenylate cyclase-activating polypeptide (PACAP)1–38 immunoreactivity, preproPACAP, and PACAP1–38 mRNA within the TNC. These increases were effectively inhibited when rats were pre-treated with KYNA, KA1, and MK-801 [63], which indicates that there is a connection between the kynurenine system and PACAP.
Notably, levels of kynurenine pathway metabolites were found altered in migraine sufferers. Decreased kynurenine metabolite levels were identified in patients with chronic migraine, cluster headache, and episodic migraine [64][65][66][67] consistent with findings from animal studies using the NTG migraine model [53]. These findings suggest that decreased KYNA levels may signify heightened glutamatergic activity in chronic migraine and cluster headache [68].
The precise role of KYNA and its metabolites in migraine pathomechanisms remains partially understood. KYNA’s effects may occur through peripheral and central mechanisms. Peripherally, KYNA can modulate glutamate receptors, particularly NMDA receptors in the dorsal root and TG [69]. Beyond peripheral effects, KYNA and analogs impact second-order neurons, as evidenced by KYNA’s reduction of mechanical allodynia and pain sensitivity in tests like the hot-plate and tail-flick tests [70][71] (Figure 3).
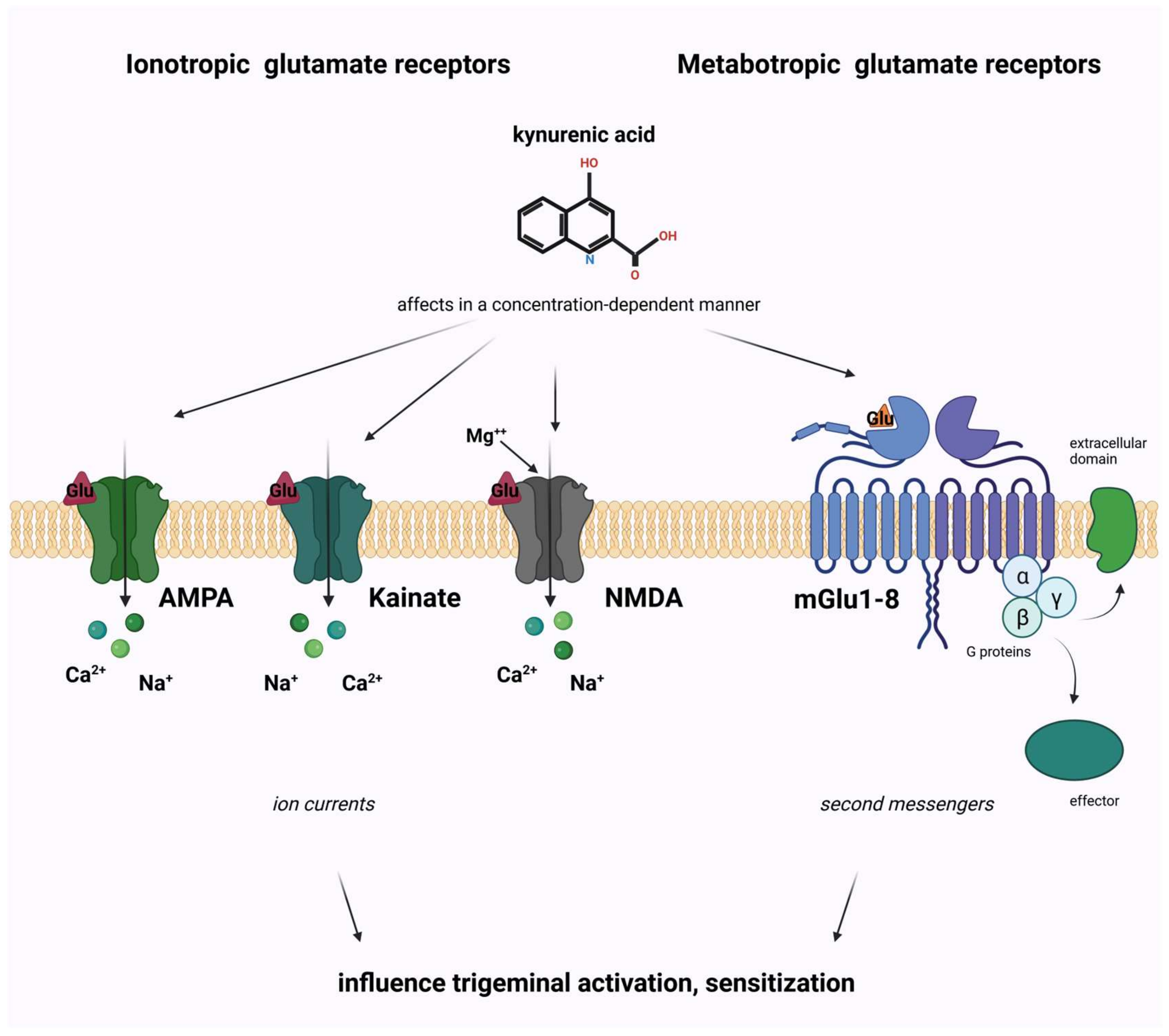
Figure 3. The role of glutamate and kynurenine system in migraine pathomechanism. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-D-aspartate; mGlu, metabotropic glutamate receptor.
References
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd ed. Cephalalgia 2013, 33, 629–808.
- Humphries, E.S.A.; Dart, C. Neuronal and Cardiovascular Potassium Channels as Therapeutic Drug Targets: Promise and Pitfalls. SLAS Discov. Adv. Sci. Drug Discov. 2015, 20, 1055–1073.
- Al-Karagholi, M.A.-M. Involvement of Potassium Channel Signalling in Migraine Pathophysiology. Pharmaceuticals 2023, 16, 438.
- Wang, J.; Ou, S.-W.; Wang, Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554.
- Südhof, T.C. Calcium Control of Neurotransmitter Release. Cold Spring Harb. Perspect. Biol. 2011, 4, a011353.
- Longden, T.A.; Hill-Eubanks, D.C.; Nelson, M.T. Ion channel networks in the control of cerebral blood flow. J. Cereb. Blood Flow Metab. 2015, 36, 492–512.
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259.
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622.
- Edvinsson, L. Tracing neural connections to pain pathways with relevance to primary headaches. Cephalalgia 2011, 31, 737–747.
- Spekker, E.; Tanaka, M.; Szabó, Á.; Vécsei, L. Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines 2021, 10, 76.
- Cross, S.A. Pathophysiology of Pain. Mayo Clin. Proc. 1994, 69, 375–383.
- Noseda, R.; Burstein, R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain 2013, 154, S44–S53.
- Dodick, D.W. A Phase-by-Phase Review of Migraine Pathophysiology. Headache J. Head Face Pain 2018, 58, 4–16.
- Pietrobon, D.; Moskowitz, M.A. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 2014, 15, 379–393.
- Zhang, X.; Levy, D.; Noseda, R.; Kainz, V.; Jakubowski, M.; Burstein, R. Activation of Meningeal Nociceptors by Cortical Spreading Depression: Implications for Migraine with Aura. J. Neursci. 2010, 30, 8807–8814.
- Gursoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455.
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of Migraine. Annu. Rev. Physiol. 2013, 75, 365–391.
- Cohen, C.F.; Roh, J.; Lee, S.H.; Park, C.-K.; Berta, T. Targeting Nociceptive Neurons and Transient Receptor Potential Channels for the Treatment of Migraine. Int. J. Mol. Sci. 2023, 24, 7897.
- Zhang, X.-C.; Strassman, A.M.; Burstein, R.; Levy, D. Sensitization and Activation of Intracranial Meningeal Nociceptors by Mast Cell Mediators. Experiment 2007, 322, 806–812.
- Levy, D. Endogenous Mechanisms Underlying the Activation and Sensitization of Meningeal Nociceptors: The Role of Immuno-Vascular Interactions and Cortical Spreading Depression. Curr. Pain Headache Rep. 2012, 16, 270–277.
- Liu, Y.; Broman, J.; Zhang, M.; Edvinsson, L. Brainstem and Thalamic Projections from a Craniovascular Sensory Nervous Centre in the Rostral Cervical Spinal Dorsal Horn of Rats. Cephalalgia 2009, 29, 935–948.
- Weiller, C.; May, A.; Limmroth, V.; Jüptner, M.; Kaube, H.; Schayck, R.; Coenen, H.; Dlener, H. Brain stem activation in spontaneous human migraine attacks. Nat. Med. 1995, 1, 658–660.
- Akerman, S.; Holland, P.R.; Goadsby, P.J. Diencephalic and brainstem mechanisms in migraine. Nat. Rev. Neurosci. 2011, 12, 570–584.
- Settle, M. The Hypothalamus. Neonatal Netw. 2000, 19, 9–14.
- Goadsby, P.J.; Holland, P.R. Pathophysiology of Migraine. Neurol. Clin. 2019, 37, 651–671.
- Strassman, A.M.; Raymond, S.A.; Burstein, R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996, 384, 560–564.
- Kullmann, D.M. The neuronal channelopathies. Brain 2002, 125, 1177–1195.
- Yan, J.; Dussor, G. Ion Channels and Migraine. Headache J. Head Face Pain 2014, 54, 619–639.
- Carrera, P.; Stenirri, S.; Ferrari, M.; Battistini, S. Familial hemiplegic migraine: A ion channel disorder. Brain Res. Bull. 2001, 56, 239–241.
- Uchitel, O.D.; Inchauspe, C.G.; Di Guilmi, M.N. Calcium channels and synaptic transmission in familial hemiplegic migraine type 1 animal models. Biophys. Rev. 2013, 6, 15–26.
- Pietrobon, D. Ion channels in migraine disorders. Curr. Opin. Physiol. 2018, 2, 98–108.
- Sutherland, H.G.; Albury, C.L.; Griffiths, L.R. Advances in genetics of migraine. J. Headache Pain 2019, 20, 72.
- Barker, B.S.; Young, G.T.; Soubrane, C.H.; Stephens, G.J.; Stevens, E.B.; Patel, M.K. Chapter 2—Ion Channels. In Conn’s Translational Neuroscience; Michael Conn, P., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 11–43.
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118.
- Alexander, S.P.; Peters, J.A.; Kelly, E.; Marrion, N.V.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. The Concise Guide to Pharmacology 2017/18: Ligand-gated ion channels. Br. J. Pharmacol. 2017, 174, S130–S159.
- Szalardy, L.; Klivenyi, P.; Zadori, D.; Fulop, F.; Toldi, J.; Vecsei, L. Mitochondrial disturbances, tryptophan metabolites and neurodegeneration: Medicinal chemistry aspects. Curr. Med. Chem. 2012, 19, 1899–1920.
- Egerton, A.; Grace, A.A.; Stone, J.; Bossong, M.G.; Sand, M.; McGuire, P. Glutamate in schizophrenia: Neurodevelopmental perspectives and drug development. Schizophr. Res. 2020, 223, 59–70.
- Henter, I.D.; Park, L.T.; Zarate, C.A. Novel Glutamatergic Modulators for the Treatment of Mood Disorders: Current Status. CNS Drugs 2021, 35, 527–543.
- Vecsei, L.; Majlath, Z.; Balog, A.; Tajti, J. Drug targets of migraine and neuropathy: Treatment of hyperexcitability. CNS Neurol. Disord. Drug Targets 2015, 14, 664–676.
- Martínez, F.; Castillo, J.; Rodríguez, J.R.; Leira, R.; Noya, M. Neuroexcitatory Amino Acid Levels in Plasma and Cerebrospinal Fluid During Migraine Attacks. Cephalalgia 1993, 13, 89–93.
- Olney, J.W.; Sharpe, L.G. Brain Lesions in an Infant Rhesus Monkey Treated with Monosodium Glutamate. Science 1969, 166, 386–388.
- Choi, D.W. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 1985, 58, 293–297.
- Koga, K.; Li, S.; Zhuo, M. Metabotropic Glutamate Receptor Dependent Cortical Plasticity in Chronic Pain. Curr. Neuropharmacol. 2016, 14, 427–434.
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur. J. Pharmacol. 1988, 154, 85–87.
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A Glycine Site Associated with N-Methyl-d-Aspartic Acid Receptors: Characterization and Identification of a New Class of Antagonists. J. Neurochem. 1989, 52, 1319–1328.
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112.
- Rózsa, Á.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091.
- Kemp, J.A.; Foster, A.C.; Leeson, P.D.; Priestley, T.; Tridgett, R.; Iversen, L.L.; Woodruff, G.N. 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex. Proc. Natl. Acad. Sci. USA 1988, 85, 6547–6550.
- Sas, K.; Robotka, H.; Rózsa, E.; Ágoston, M.; Szénási, G.; Gigler, G.; Marosi, M.; Kis, Z.; Farkas, T.; Vécsei, L.; et al. Kynurenine diminishes the ischemia-induced histological and electrophysiological deficits in the rat hippocampus. Neurobiol. Dis. 2008, 32, 302–308.
- Fejes-Szabó, A.; Bohár, Z.; Vámos, E.; Nagy-Grócz, G.; Tar, L.; Veres, G.; Zádori, D.; Szentirmai, M.; Tajti, J.; Szatmári, I.; et al. Pre-treatment with new kynurenic acid amide dose-dependently prevents the nitroglycerine-induced neuronal activation and sensitization in cervical part of trigemino-cervical complex. J. Neural Transm. 2014, 121, 725–738.
- Vamos, E.; Pardutz, A.; Fejes, A.; Tajti, J.; Toldi, J.; Vecsei, L. Modulatory effects of probenecid on the nitroglycerin-induced changes in the rat caudal trigeminal nucleus. Eur. J. Pharmacol. 2009, 621, 33–37.
- Vámos, E.; Párdutz, Á.; Varga, H.; Bohár, Z.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L. l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology 2009, 57, 425–429.
- Nagy-Grócz, G.; Tar, L.; Bohár, Z.; Fejes-Szabó, A.; Laborc, K.F.; Spekker, E.; Vécsei, L.; Párdutz, Á. The modulatory effect of anandamide on nitroglycerin-induced sensitization in the trigeminal system of the rat. Cephalalgia 2015, 36, 849–861.
- Nagy-Grócz, G.; Laborc, K.F.; Veres, G.; Bajtai, A.; Bohár, Z.; Zádori, D.; Fejes-Szabó, A.; Spekker, E.; Vécsei, L.; Párdutz, Á. The Effect of Systemic Nitroglycerin Administration on the Kynurenine Pathway in the Rat. Front. Neurol. 2017, 8, 278.
- Lukács, M.; Warfvinge, K.; Kruse, L.S.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L.; Edvinsson, L. KYNA analogue SZR72 modifies CFA-induced dural inflammation- regarding expression of pERK1/2 and IL-1β in the rat trigeminal ganglion. J. Headache Pain 2016, 17, 64.
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17.
- Cseh, E.K.; Veres, G.; Körtési, T.; Polyák, H.; Nánási, N.; Tajti, J.; Párdutz, Á.; Klivényi, P.; Vécsei, L.; Zádori, D. Neurotransmitter and tryptophan metabolite concentration changes in the complete Freund’s adjuvant model of orofacial pain. J. Headache Pain 2020, 21, 35.
- Fejes-Szabo, A.; Bohar, Z.; Nagy-Grocz, G.; Vamos, E.; Tar, L.; Podor, B.; Tajti, J.; Toldi, J.; Vecsei, L.; Pardutz, Á. Effect of probenecid on the pain-related behaviour and morphological markers in orofacial formalin test of the rat. CNS Neurol. Disord. Drug Targets 2015, 14, 350–359.
- Veres, G.; Fejes-Szabó, A.; Zádori, D.; Nagy-Grócz, G.; László, A.M.; Bajtai, A.; Mándity, I.; Szentirmai, M.; Bohár, Z.; Laborc, K.; et al. A comparative assessment of two kynurenic acid analogs in the formalin model of trigeminal activation: A behavioral, immunohistochemical and pharmacokinetic study. J. Neural Transm. 2016, 124, 99–112.
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Redavide, E.; Pampalone, S.; Toldi, J.; Fülöp, F.; Blandini, F.; Nappi, G.; Sandrini, G.; et al. Effects of kynurenic acid analogue 1 (KYNA-A1) in nitroglycerin-induced hyperalgesia: Targets and anti-migraine mechanisms. Cephalalgia 2016, 37, 1272–1284.
- Knyihár-Csillik, E.; Chadaide, Z.; Okuno, E.; Krisztin-Péva, B.; Toldi, J.; Varga, C.; Molnár, A.; Csillik, B.; Vécsei, L. Kynurenine aminotransferase in the supratentorial dura mater of the rat: Effect of stimulation of the trigeminal ganglion. Exp. Neurol. 2004, 186, 242–247.
- Knapp, L.; Szita, B.; Kocsis, K.; Vécsei, L.; Toldi, J. Nitroglycerin enhances the propagation of cortical spreading depression: Comparative studies with sumatriptan and novel kynurenic acid analogues. Drug Des. Dev. Ther. 2016, 11, 27–34.
- Körtési, T.; Tuka, B.; Tajti, J.; Bagoly, T.; Fülöp, F.; Helyes, Z.; Vécsei, L. Kynurenic Acid Inhibits the Electrical Stimulation Induced Elevated Pituitary Adenylate Cyclase-Activating Polypeptide Expression in the TNC. Front. Neurol. 2018, 8, 745.
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47.
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2015, 17, 27.
- Tuka, B.; Nyári, A.; Cseh, E.K.; Körtési, T.; Veréb, D.; Tömösi, F.; Kecskeméti, G.; Janáky, T.; Tajti, J.; Vécsei, L. Clinical relevance of depressed kynurenine pathway in episodic migraine patients: Potential prognostic markers in the peripheral plasma during the interictal period. J. Headache Pain 2021, 22, 60.
- Tuka, B.; Körtési, T.; Nánási, N.; Tömösi, F.; Janáky, T.; Veréb, D.; Szok, D.; Tajti, J.; Vécsei, L. Cluster headache and kynurenines. J. Headache Pain 2023, 24, 35.
- Curto, M.; Lionetto, L.; Fazio, F.; Mitsikostas, D.-D.; Martelletti, P. Fathoming the kynurenine pathway in migraine: Why understanding the enzymatic cascades is still critically important. Intern. Emerg. Med. 2015, 10, 413–421.
- Sato, K.; Kiyama, H.; Park, H.T.; Tohyama, M. AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. NeuroReport 1993, 4, 1263–1265.
- Mecs, L.; Tuboly, G.; Nagy, E.; Benedek, G.; Horvath, G. The Peripheral Antinociceptive Effects of Endomorphin-1 and Kynurenic Acid in the Rat Inflamed Joint Model. Obstet. Anesthesia Dig. 2009, 109, 1297–1304.
- Zhang, Y.-Q.; Ji, G.-C.; Wu, G.-C.; Zhao, Z.-Q. Kynurenic acid enhances electroacupuncture analgesia in normal and carrageenan-injected rats. Brain Res. 2003, 966, 300–307.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
06 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No