Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dmitry D. Zhdanov | -- | 6762 | 2023-11-22 09:07:15 | | | |
| 2 | Lindsay Dong | Meta information modification | 6762 | 2023-11-23 08:43:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shishparenok, A.N.; Gladilina, Y.A.; Zhdanov, D.D. Engineering and Expression Strategies for Optimization of L-Asparaginase. Encyclopedia. Available online: https://encyclopedia.pub/entry/51898 (accessed on 26 July 2026).
Shishparenok AN, Gladilina YA, Zhdanov DD. Engineering and Expression Strategies for Optimization of L-Asparaginase. Encyclopedia. Available at: https://encyclopedia.pub/entry/51898. Accessed July 26, 2026.
Shishparenok, Anastasiya N., Yulia A. Gladilina, Dmitry D. Zhdanov. "Engineering and Expression Strategies for Optimization of L-Asparaginase" Encyclopedia, https://encyclopedia.pub/entry/51898 (accessed July 26, 2026).
Shishparenok, A.N., Gladilina, Y.A., & Zhdanov, D.D. (2023, November 22). Engineering and Expression Strategies for Optimization of L-Asparaginase. In Encyclopedia. https://encyclopedia.pub/entry/51898
Shishparenok, Anastasiya N., et al. "Engineering and Expression Strategies for Optimization of L-Asparaginase." Encyclopedia. Web. 22 November, 2023.
Copy Citation
L-asparaginase, an enzyme widely used in the clinic for the treatment of leukemia and in bakeries for the reduction of acrylamide. Newly developed recombinant L-asparaginase (L-ASNase) may have a low affinity for asparagine, reduced catalytic activity, low stability, and increased glutaminase activity or immunogenicity. Some successful commercial preparations of L-ASNase are now available. Therefore, obtaining novel L-ASNases with improved properties suitable for food or clinical applications remains a challenge.
L-asparaginase
directed evolution
rational design
heterologous expression
genetic engineering
1. Introduction
L-asparaginase (L-ASNase, E.C.3.5.1.1) hydrolyzes asparagine to produce ammonia and L-aspartic acid. The enzyme is widely used in the pharmaceutical and food industries. L-ASNase is a key therapeutic component in the treatment of lymphosarcoma and acute lymphoblastic leukemia (ALL) [1][2][3][4][5]. Recent research has shown that L-ASNase has clinical potential in the treatment of several aggressive subtypes of hematological or solid tumors, including glioblastoma, breast, pancreatic and hepatocellular carcinomas [6][7]. The anticancer effect of L-ASNase is based on its ability to hydrolyze L-asparagine, which is necessary for neoplastic cells. Leukemic cells cannot synthesize this amino acid due to the absence or lack of L-asparagine synthetase and depend on the exogenous supply from the bloodstream. Depletion of asparagine leads to impaired protein synthesis and starvation of cancer cells, leading to cell death [1]. The second view, or non-canonical approach, is that L-ASNase acts directly on cancer cells. The effects of ASNase on ROS levels, cell cycle progression, autophagy and apoptotic cell death have been demonstrated. Another anti-cancer strategy under L-ASNase treatment is the inhibition of the Akt/mTOR and Erk pathways [8]. L-ASNase is also a promising agent for the food industry because it reduces the formation of toxic acrylamide. Acrylamide is formed as a result of the nonenzymatic interaction of sugars with L-asparagine when starchy foods are heated to 120 °C under low humidity conditions in the Maillard reaction [9][10][11]. Effects unrelated to the hydrolysis of asparagine or glutamine have been described for a number of L-ASNases [12][13][14].
2. Host Systems for Expression of L-Asparaginase
L-ASNases are widespread in nature [15]. According to the National Centre for Biotechnology Information (NCBI), L-ASNase sequences are mostly found in bacteria, accounting for 95.5% of all protein sequences deposited (221,303 out of 231,770). However, L-ASNAse can also be found in the fungi (1.68%), animal (1.25%), plant (0.24%), archaea (0.88%) and kingdoms [16].
The best producers of L-ASNase belong to the family Enterobacteriaceae, followed by species of fungi. The main hosts for L-ASNase expression systems to date are E. coli, Bacillus subtilis (B. subtilis) and Pichia pastoris (P. pastoris) [17]. Among bacteria, wild types of Bacillus are natural producers of L-ASNase, e.g., Bacillus australimaris NJB19 (MG734654) [18] and Bacillus lichenformis [19].
L-ASNases have been discovered producing enzymes in eukaryotes, such as fungi and yeasts, that have fewer negative consequences and greater beneficial properties [20]. Eukaryotic systems offer several advantages such as proper folding, efficient secretion, and typical eukaryotic post-translational modifications [20][21][22]. A number of yeast genera with considerable L-ASNase production capacity have been documented, mainly from the genera Candida, Yarrowia and Rhodotorula, whereas Aspergillus, Penicillium and Fusarium dominate the literature for L-ASNase-producing mold genera [23][24]. Although fungal L-ASNases are often produced extracellularly, which facilitates downstream purification [25], S. cerevisiae strains were found to produce both intracellular and extracellular forms of L-ASNase. Blue-green microalgae is an alternative source of L-ASNase that is attractive due to its lack of seasonal variation, low cost of media formulation, and ease of growth and harvest [26].
Human cells were also used to express a recombinant bacterial L-ASNase. HEK293 showed highly comparable relative activity profiles with L-ASNase produced in E. coli at different pH conditions and temperatures [27]. Recombinant human L-ASNase (ASRGL1) was obtained in Baculovirus and mammalian cells and can also be purchased from the MyBioSource company [28].
However, microorganisms such as bacteria, filamentous fungi and yeast are considered to be the best source of L-ASNase due to their ability to grow rapidly on extremely simple and inexpensive substrates. The production of L-ASNase can also be easily genetically modified, depending on the strain used, making the extraction and purification process commercially viable [20]. Additionally, the biotechnological production process is usually easier to optimize and scale-up than other processes [23]. Examples of heterologous expression of L-ASNases in various hosts are described in detail by Castro et al. [29].
L-ASNases are currently classified into three major groups: bacterial type L-ASNase (Class 1, containing classification Types I and II), plant type L-ASNase (Class 2, Type III), and rhizobial type L-ASNase (Class 3) [29][30] Cytosolic bacterial type I L-ASNases are involved in nitrogen metabolism and appear to be expressed constitutively [31]. In mesophiles, these enzymes exhibit low affinity towards L-asparagine with KM values in the millimolar range. Periplasmic bacterial type II L-ASNases appear to participate in carbon metabolism and their expression is tightly regulated by different factors [32]. Type II enzymes of mesophilic origin exhibit a high specific activity towards L-asparagine with micromolar KM. Plant-type L-ASNases have dual L-asparaginase and isoaspartyl aminopeptidase activity. These enzymes are located in the periplasmic space and hydrolyze the side-chain amide bond of L-asparagine or its β-peptides [32]. They are low-affinity proteins (millimolar KM) that belong to the superfamily of N-terminal nucleophile hydrolases [33][34].
Based on structural and kinetic data, two general catalytic mechanisms have been proposed for Class 1 L-ASNases (Type I, Type II): a single displacement (or direct displacement) mechanism for mammalian enzymes and a double displacement (ping-pong) mechanism for bacterial enzymes [35], which is thought to proceed via a covalent acyl-enzyme intermediate. In Class 1 enzymes (bacterial type), the N-terminal domain has all the elements necessary for catalysis [31]. Type II L-ASNases have a molecular weight that varies between 31.0 and 42.0 kDa [36]. Most marine L-ASNases from different sources have molecular masses in the range of 25–41.1 kDa, with the exception of Bacillus pumilus L-ASNase, which is active as a dimer or tetramer with a molecular mass of 71 kDa [30]. L-ASNases of the plant type (Type III, Class 2) are found in microorganisms, insects and mammals, including humans. They are Ntn hydrolases with dual isoaspartyl aminopeptidase/l-asparaginase activity (EC 3.5.1.1/3.4.19.5). Human HsAIII and E. coli EcAIII are the most widely studied class 2 L-ASNases [31].
Some of the structural features of an L-ASNase protomer vary slightly between individual enzymes; nevertheless, the overall topology appears to be very highly conserved. The biological assembly of both type I and II ASNases is a homotetramer [37][38]. The whole assembly is relatively compact. The tetramer can be represented as a dimer consisting of two intimate dimers. The interface between protomers within such a small dimer is larger than the interface between protomers from different dimers. Each dimer forms two complete active sites, raising the question of the role of tetramerization. As there are four active sites at the interface of two subunits forming an intimate dimer, L-ASNase is more accurately characterized as a dimer of dimers with an anticancer molecular mass of around 120 to 160 kDa [39]. The active sites of L-ASNases are located at the interfaces of intimate dimers, with each intimate dimer having two active site pockets generated by amino acids from both subunits. Structural and functional studies have shown that the catalytic triad consisting of three polar amino acids, Thr-Lys-Asp (Thr89, Lys162 and Asp90 in EcA), is required for enzyme activity [40]. Investigation of the structure of L-ASNase with ligand molecules in the active site revealed the formation of an intricate hydrogen network with ligands and the discovery of two new residues important for the catalytic mechanism (Thr12 and Tyr25 in EcA) [32]. These residues are located in a broad loop (amino acids 10–32 in EcA) that acts as a lid for the active site, most likely facilitating substrate binding and thus catalysis [23].
In industrial production, L-ASNase can be secreted intracellularly so that cell destruction occurs in several stages. To overcome this problem, a variety of molecular tools and techniques are available for high-level expression of heterologous L-ASNases, including the design of expression plasmids, engineered production hosts, and growth and culture strategies [41]. The use of both naturally occurring hyperproductive strains and those created through genetic engineering, in particular CRISPR/Cas9 systems, protoplast fusion, and response surface methodology, as well as the design of experiments using artificial neural networks and response surfaces, has significantly increased yields [42][43]
2.1. Escherichia coli Expression System
L-ASNase I and L-ASNase II are the two different forms of L-ASNases that have been found in E. coli [44]. Among these, L-ASNase II was the primary source of commercially used L-ASNase [17]. Asparaginase isozyme II (AnsB) from E. coli is a prokaryotic protein that has been studied for the treatment of acute lymphoblastic leukemia (ALL) for more than 50 years. Much research has been devoted to the active expression of AnsB in E. coli [45]. The advantages of using E. coli as a host are well-known: fast growth kinetics, high cell densities, easily available and inexpensive components of media and easy and rapid transformation of exogenous plasmid DNA [46]. However, the E. coli expression system has some drawbacks, including a lack of eukaryotic post-translational modifications, limited solubility, inappropriate protein folding, inclusion body formation, endotoxins and inadequate secretion [47].
Expression vectors useful for the production of L-ASNase include vectors derived from bacterial plasmids, bacteriophage, yeast episome, yeast chromosomal elements, viruses such as baculovirus, papovavirus, vaccinia virus, adenovirus, fowl pox virus, pseudorabies virus and retroviruses, and vectors derived from combinations thereof, such as those derived from plasmid [48].
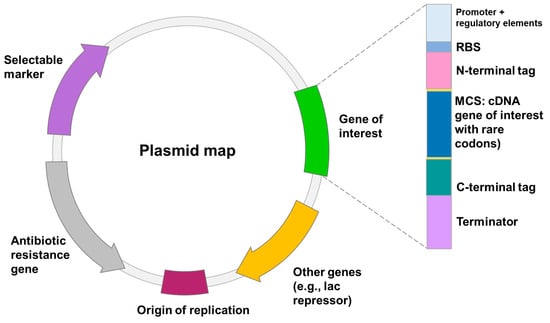
Heterologous gene transcription must be optimized to avoid these shortcomings and to produce competitive biotechnological processes [49]. Today, a wide range of cutting-edge strategies and high-throughput techniques are used to improve recombinant protein expression in E. coli. These include strong promoters, novel expression systems, protein tags, codon optimization, secretion signals, E. coli engineering for disulfide bond formation and glycoengineering, co-expression of foldases and molecular chaperons, metabolic engineering, high-throughput cloning and screening tools, and fermentation technologies [41]. Elements on the plasmid to optimize heterologous expression are shown in Figure 1.
Figure 1. The schematic of the transcription elements on the plasmid to optimize expression of recombinant proteins in E. coli and B. subtilis. These elements include: (1) origin of replication (ori); (2) translation initiation sequence: a ribosomal-binding site (RBS) and start codon; (3) promoter; (4) affinity tag; (5) multiple cloning site (MCS); (6) transcription termination sequence; (7) selectable marker.
The most common method to produce recombinant proteins is the use of pET vectors in E. coli BL21(DE3) hosts [50]. E. coli BL21 (DE3) may also be a favorable host for the production of recombinant proteins, e.g., L-ASNase, because it lacks the cytoplasmic protease Lon and the outer membrane protease OmpT, which can increase the stability of a produced recombinant protein [51][52]. OmpT is best avoided as it is a relatively stable protease that can damage endogenous and recombinant proteins after cell lysis. All of these features combine to make E. coli BL21(DE3) a suitable host for recombinant protein production in the cytoplasm and cell envelope, and E. coli BL21(DE3) and its variants dominate protein production [53].
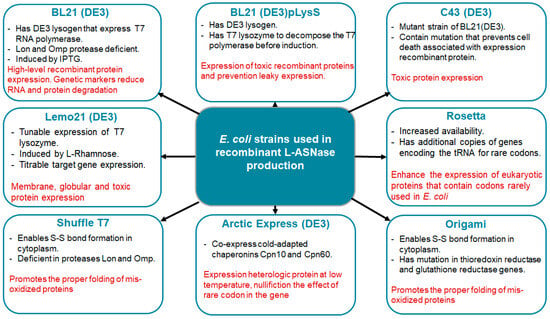
Several E. coli strains (Figure 2) developed from BL21(DE3) are currently commercially available, each with unique properties to enhance protein expression. The strains represent modifications such as chaperonin co-expression (ArcticExpress(DE3) and GroEL(DE3)), tight expression control (Turner(DE3), pLysS (DE3) and Lemo21(DE3)), cytosolic pro-oxidant environment (Origami B (DE3) and Shuffle T7) and specialization in toxic protein expression (C43(DE3)). Despite the commercial interest in ASNases, only a few strains have been used for enzyme production, the most common being BL21 (DE3). Its protein was produced without a signal peptide for periplasmic export [54].
Figure 2. The most popular commercial E. coli strains to avoid potential pitfalls of recombinant L-ASNase expression. The key characteristics of the strains are highlighted in black. The advantages are highlighted in red.
De Moura et al. tested eleven different E. coli expression strains to determine the one best suited for the expression of cytosolic L-ASNase at 16 °C using wild-type recombinant EcAII (rEcAII) with a His-tag as the prototype enzyme. BL21 ArcticExpress(DE3) showed the best results among the E. coli strains examined, with reduced protein aggregates, correct folding and higher specific activity (156 U/mg), indicating that this strain is perfectly suited for recombinant L-ASNase development [55].
2.2. Bacillus subtilis Expression System
B. subtilis is a nonpathogenic and GRAS (generally regarded as safe) bacterium. The main advantage of B. subtilis is that it does not produce LPS, which can cause degenerative diseases in humans and animals. Because of its genetic characteristics, B. subtilis can also be easily modified using a variety of bacteriophages and plasmids. It can secrete functional extracellular proteins directly into the culture medium, facilitating subsequent purification steps. It has no major bias in codon usage, which is considered an advantage [56]. Compared to the E. coli expression system, B. subtilis has negligible protein secretion potential, and little information is available on the production of L-ASNases by B. subtilis [17].
There are many strains of B. subtilis, including auxotrophic strains 23, 122, 160, 166 and 168, which were initially developed from B. subtilis. The 168 strain has been widely employed in academic and industrial research. To obtain mutant strains that meet production needs, several strains can be altered through systematic metabolic engineering. For the production of heterologous proteins, B. subtilis WB600 and WB800 strains were created by the removal of six and eight protease genes, respectively, from B. subtilis 168 [57].
Expression systems for the production of recombinant proteins in B. subtilis can be created using either independent plasmid expression or expression from a chromosome-integrated expression cassette. The B. subtilis expression system has some disadvantages compared to the E. coli expression system, including unstable plasmids, lack of plasmid diversity and poor plasmid transformation rates. Plasmid stability and copy number can usually be altered by introducing a replication gene [58]. There are few standardized promoters documented in the B. subtilis registry compared to E. coli, and most are constitutive elements [59]. Insertion of the ori region has been shown to promote plasmid replication in B. subtilis [60]. Expression plasmids with improved structural stability have also been generated from θ-replication plasmids [61]. When using single plasmids to express recombinant proteins, multicopy plasmids are required [62].
Several expression systems have been developed in different Bacillus species to overproduce heterologous proteins [63]. Constitutive promoters, double promoters, functional synthetics capable of directing transcription, effective signaling peptides, inducible and self-inducing expression systems and post-transcription regulation can improve the expression level of recombinant proteins in B. subtilis [57][58].
2.3. Pichia pastoris Expression System
P. pastoris is an effective platform for the production of high-titer recombinant proteins. P. pastoris is a methylotrophic yeast typically grown using dynamic culture methods. It has certain unique physiological characteristics, such as the ability to grow rapidly at high cell densities in basal media and to produce proteins in high yields under bioreactor conditions.
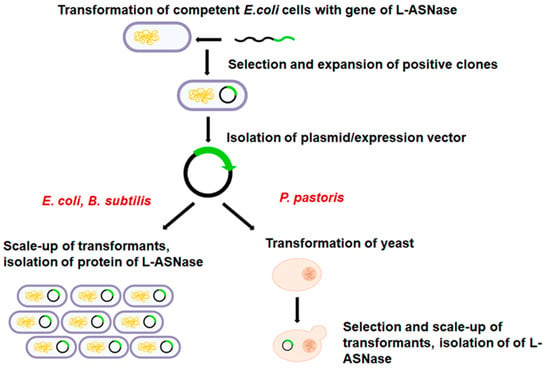
The advantages of protein production using the P. pastoris system include improved folding efficiency, high cell density fermentation, a powerful expression system, genetic stability and a mature secretion system for secreting proteins into the external environment [64]. In contrast to bacteria, linearized foreign DNA can be efficiently incorporated into a chromosome by a process called cross-recombination, to create stable yeast cell lines (Figure 3) [65]. Furthermore, P. pastoris is a suitable bacterium for the secretory synthesis of recombinant proteins directly into the culture medium supernatant. Due to the minimal synthesis of endogenous secretory proteins in the P. pastoris expression system, the purification of recombinant proteins is straightforward [66]. P. pastoris, like Saccharomyces, benefits from molecular and genetic engineering, but the expression levels of heterologous proteins are 10 to 100 times higher, making P. pastoris more popular [67]. However, P. pastoris, unlike S. cerevisiae, is not a fermentative yeast. As a result, almost all glucose is converted to biomass. Because glucose is not converted to ethanol or other organic acids, P. pastoris can grow to high cell densities under aerobic conditions [68].
Figure 3. The following are the critical steps in the production of heterologous L-ASNase in P. pastoris: (1) the L-ASNase gene (cDNA) is extracted from sample and inserted into the host vector; (2) E. coli is commonly used to amplify the constructs; (3) yeast cells are transformed with a vector encoding the cDNA for the L-ASNase gene and colonies are grown on selective medium. This step is different in bacteria.
A variety of P. pastoris expression vectors and host strains are available [69]. Commonly used P. pastoris vectors for the expression of heterologous proteins are pGAPZ, pHIL-S1, pPIC9K, pJL-SX, pBLHIS-SX, pPICZ, pHIL-D2, pJL-IX and pBLHIS-IX. Expression vectors in P. pastoris, such as shuttle vectors in E. coli, can be transferred between two different host species. In addition, the vectors contain a drug resistance gene, such as Kan, Shble, Bsd, Amp or FLD1, which is resistant to geneticin, zeocin, blasticidin, ampicillin and formaldehyde [66]. The P. pastoris strain GS115 is a commonly used strain for protein expression. It has two genes encoding alcohol oxidase enzymes, AOX1 and AOX2. This yeast first attracted attention for its inherent ability to use methanol as its sole carbon supply, which can be achieved through specific metabolic pathways involving numerous specialized enzymes. Over 5000 proteins have been successfully cloned and produced using the P. pastoris method. However, P. pastoris is unable to synthesize or secrete all the proteins in such high concentrations. Protein production is significantly reduced under normal conditions, especially when complex hetero-oligomeric proteins are produced [64]. The KM71 strain is a derivative of the GS115 strain and has the AOX1 gene deleted, making it a MutS strain [70]. The X33 strain is a prototrophic strain, which means that it can grow on minimal media without the need for any additional nutrients. The X33 strain is Mut+, which means that it is able to use methanol as a carbon source and has a functional AOX1 gene [71]. Other strains, such as SMD1163 (his4 pep4 prb1), SMD1165 (his4 pep4), SMD1168 (his4 pep4), BG21 and PichiaPinkTM, have no protease, which prevents the degradation of a secreted protein [72].
For intracellular and secretory expression, Life Technologies offers a range of standard P. pastoris expression vectors with constitutive (PGAP) and inducible promoters (PAOX1, PFLD). By using ade2 knockout strains and truncated ADE2 promoters of different intensities in front of the ADE2 marker gene, the PichiaPinkTM expression kit for intracellular or secretory expression facilitates the selection of multicopy integration clones. BioGrammatics also offers GlycoSwitch® vectors designed for humanized glycosylation of target proteins and holds licenses for the sale of standard P. pastoris expression vectors and strains [71]. Some commonly employed Pichia GlycoSwitch® strains are SuperMan5, SuperMan5HIS−, SuperMan5pep4–, and SuperMan5(aox1–, Muts) [66].
In P. pastoris, selectable markers are used to identify cells that have taken up the recombinant DNA and to maintain the recombinant DNA in the cells during growth and division. The commonly used selectable markers in P. pastoris include: (1) HIS4: the HIS4 gene is a histidine biosynthesis gene that is used as a selectable marker in the transformation of P. pastoris cells; (2) URA3 and URA5: the URA3 and URA5 genes are also often used as selection markers in P. pastoris (3) ADE1, ARG4 and MET2: these genes are also used as selectable markers in P. pastoris. The choice of selectable marker depends on the specific application and the availability of the appropriate auxotrophic strains [73][74].
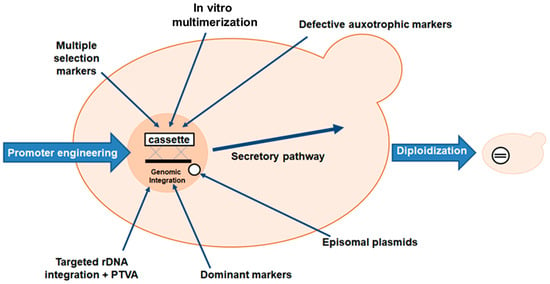
Many researchers have used molecular techniques to increase protein production due to the low specific productivity of P. pastoris. The most common method of achieving this goal is to deliver increased titers of a particular transcript to the cell’s translation machinery. This is usually achieved by using a suitable promoter to stimulate production of the heterologous gene or simply by increasing the copy number of the target gene (Figure 4) [75]. Pan et al. also described some other approaches, such as host strain engineering by improving homologous recombination efficiency, using episomal plasmids for heterologous gene expression and the CRISPR/Cas9 system for genome editing, selective marker and marker recycling and metabolic engineering [64]. Codon optimization, introduction of artificial glycosylation sites, signal peptide engineering, and cell surface display have also been suggested [70].
Figure 4. Strategies used in P. pastoris to enhance the transcription of the gene of interest (L-ASNase). The titer of heterologous transcripts can be increased by: (1) promoter engineering; (2) introduction of multiple copies of the heterologous gene to obtain multi-copy clones; (3) in vitro construction of multimers of the expression cassette to obtain multi-copy clones; (4) use of dominant markers in multiple clones to increase drug resistance. The use of dominant markers is the basis of the post-transformational vector amplification (PTVA) method; (5) use of defective auxotrophic markers; (6) diploidization of selected clones; (7) combinatorial engineering of secretion helpers involved in protein folding and vesicle trafficking.
One of strategies to enhance the transcription of the gene of interest (Figure 4), in which gene expression cassettes can be incorporated multiple times into the genome, was used for the production of L-ASNase. L-ASNase (GenBank accession number KF290772.1) from R. miehei was provided as a component of the widely used pCC1 cloning vector and generated in P. pastoris. Six clones carrying different numbers of gene copies (1–5 and 5+) were used to study the effect of gene copy number on the increase in protein production to increase the amount of L-ASNase. According to the findings, the clone with three integrated copies of the expression cassette had the maximum production level [76]. Strategies used to enhance L-ASNase production in P. pastoris include promoter engineering, fusion tags, signaling sequences, metabolic engineering and optimization of culture conditions.
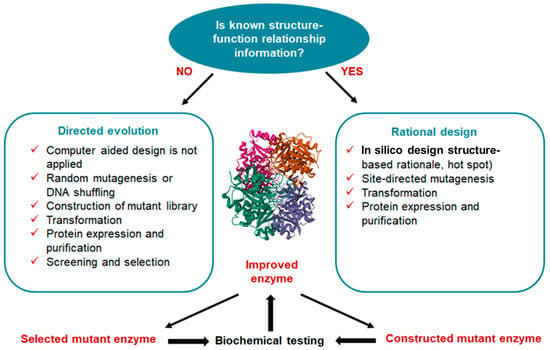
3. Approaches for Engineering Novel L-ASNases
There are two typical approaches to the molecular modification of enzymes: directed evolution and rational design (Figure 5) [77]. Combinatorial techniques (e.g., directed evolution) based on random mutation have proven effective in repurposing existing proteins to take on new roles and in generating new catalytic activities from random sequences. However, because there is so much sequence space to explore in a design challenge, certain practical choices must be made to reduce the search space to a feasible size. It is therefore difficult, if not impossible, to make a sharp distinction between rational and combinatorial techniques. Understanding the molecular basis of the protein feature that is the focus of the design study (structure–function–energetics relationship) is a fundamental requirement of rational design [78].
Figure 5. Comparison of directed evolution and rational design to the molecular modification of enzymes. Directed evolution is an approach that involves generating genetic diversity and selecting for improved variants to produce a protein with improved characteristics. In contrast to rational methods, directed evolution generates random mutations in the gene of interest and does not require any information about the protein structure. Rational design emphasizes an early understanding of protein structure and amino acid relationships. Rational design uses site-directed mutagenesis and computer modelling tools. In the center is unmodified L-ASNase from E. coli. (Protein Data Bank data source 6V5F. The structures were visualized using the PyMOL program; Schrodinger Inc., New York, NY, USA).
It is essential to be able to predict the type and position of mutations that will increase enzyme activity. Recruitment and enhancement of pre-existing activity in the protein scaffold are key components of the current paradigm of enzyme evolution. Many enzymes exhibit promiscuous activity with respect to secondary substrates in addition to high catalytic efficiency with respect to their native substrate. The idea that this latent promiscuous behavior serves as a reservoir for improving the functionality of enzymes has received much attention. Much research in recent years has identified genetic changes that are associated with increased activity during evolutionary cycles. Directed evolution and bioinformatics-based methods can be used to optimize enzyme activity. While directed evolution and bioinformatical research have revealed the contributions of both proximal and distal mutations, these studies are more biased in that they represent only mutations that were selected during adaptation. Although there are several mutations that increase activity > 10 Å from the catalytic center, in general, mutations closer to the active site (<10 Å) are likely to have a greater effect. Distal mutations can also lead to significant increases in activity. The overall improvement in activity relative to the total number of mutations varies significantly depending on the enzyme model, according to a new comprehensive review of directed evolution research [79].
4. Combining Computer Design Techniques to Obtain L-Asparaginases with Improved Properties
4.1. Modifications to Increase the Expression
Recent research indicates that rare codons are critical for protein activity and folding. However, substitution of these rare codons can lead to protein expression problems due to premature translation termination and pausing. Protein expression problems may be resolved by paying close attention to ribosomal pausing codons. Mortazavi et al. studied enzyme properties such as CUP (codon usage preference) and a rare codon cluster (RCC) in L-ASNase II Pfam PF00710 in silico. By modeling the L-ASNase 3D structure, some rare codons were discovered that may play a crucial role in the structural and functional properties of the enzyme. It is well known that changes in CUP lead to a number of problems caused by heterologous gene expression. The characteristics of the substrate-binding sites were then investigated using the molecular docking approach. AutoDock Vina was used to determine the substrate-binding sites. Several amino acids with structural similarities to the substrate-binding site of L-ASNase (2jk0) were identified. The Swiss Model and I-TASSER web server were used to model the 3D structure of the L-ASNase II enzyme. The Sherlock program was used to study its rare codons. GCUA tools (http://gcua.schoedl.de/seqoverall_v2.html (accessed on 11 October 2023)) were used to investigate the diversity in CUP of the L-ASNase II gene. Five unusual rare codons, Lys195, Leu30, Lys184, Lys160 and Lys174, were found and examined in the structure of L-ASNase. Substrate binding studies revealed that Asp6, Asp77 and Thr78 bind to the active site of the enzyme. The results of this work may contribute to a better understanding of the folding and expression of L-ASNase [80].
4.2. Modifications to Decrease Glutaminase Activity
The dual activity of L-ASNase and L-glutaminase in bacterial L-ASNases makes therapeutic use difficult. As L-asparagine is found at approximately 50 μM in human blood, therapeutic L-ASNase must have a low micromolar substrate affinity. A low Km coincides with a high kcat, ensuring that therapeutic L-ASNase effectively reduces endogenous L-asparagine at safe doses [81]. The percentage of glutaminase activity associated with L-ASNase should also be low, with excellent enzyme stability and half-life to make it an ideal therapeutic formulation [82].
Understanding the key features of L-ASNase substrate selectivity is a necessary step toward the development of less toxic enzyme variants. Several ways to reduce L-ASNase toxicity have been investigated, including mutagenic modification of the protein structure, generation of mutants with reduced ability to hydrolyze L-glutamine, chemical modification of specific amino acids, and changes in drug formulations [83]. An in-depth knowledge of the molecular basis controlling the selectivity between L-asparagine and L-glutamine will greatly benefit the rational design of L-ASNase variants with reduced L-glutaminase activity [84]. To achieve the reduction of L-glutaminase, a combination of computational protein engineering tools was used.
The crystal structure of E. chrysantemi L-ASNase in combination with glutamine was revealed and compared with the previously published complex of E. chrysantemi L-ASNase with asparagine by Nguyen et al. They concluded that to design variations that discriminate against glutamine, it is necessary to introduce mutations that are incompatible with the closed state of the enzyme when binding L-glutamine but remain compatible with the closed state when binding L-asparagine [84]. Then, rational design was used to obtain new mutant variants of E. chrysanthemi L-ASNase with reduced L-glutaminase activity. The main strategy was to search for non-conserved residues surrounding the active site that do not necessarily interact with the substrate. Based on MSA and crystal structure analysis of ErA in complex with Asp and Glu products, amino acid residues Ala-31, Glu-63, Pro-123 and Ser-254 were identified as four potential candidate sites for mutagenesis. The ErA triple mutant A31I/E63Q/S254Q (ErA-TM2) showed the optimal combination of high L-asparaginase/low L-glutaminase properties. When the l-glutaminase activity of ErA-TM2 was examined, a dramatic change in its activity profile became apparent. ErA-TM2, a derivative of ErA-WT, a somewhat active L-glutaminase, was transformed into an enzyme with extremely low L-glutaminase activity. Its rate was reduced more than 1000-fold at relevant glutamine concentrations. In vitro, an ultralow ErA-TM2 mutant effectively killed human T-ALL LOUCY cells with fewer side effects [85].
The glutaminase-deficient mutant E. coli L-ASNase was efficiently designed using molecular dynamics modeling and site-saturation mutagenesis. Molecular dynamics simulations of clinically standard E. coli L-ASNase were used to predict the structures of mutant forms that retain asparagine but not glutamine activity. Modeling was performed in the NPT ensemble using the NAMD2 package with CHARMM force field parameters and grid-based CMAP correction. Residues that reacted preferentially with glutamine rather than asparagine but were not essential for enzymatic conversion were selected as candidates for saturation mutagenesis. The best candidate was amino acid Q59. The mutant (Q59L) showed the lowest percentage of original glutaminase activity but retained >60% of wild-type (WT) asparaginase activity. The authors also found that Q59L exhibited selective anticancer activity against L-ASNase-negative leukemia cell lines, while the WT enzyme exhibited L-ASNase-independent toxicity [86].
L-ASNase from Pectobacterium carotovorum was engineered in silico to minimize glutaminase side activity for efficient treatment of ALL. In silico mutagenesis was used to generate enzyme variants with reduced glutaminase activity by changing amino acids near the ligand-binding region. Replacing the active site amino acid of the Asp96 enzyme with alanine reduced glutaminase activity by 30% while increasing asparaginase activity by 40%. Docking experiments were carried out using AutoDock 4.0, and it was found that the binding energy of the native enzyme when docked with glutamine is −8.08 kcal/mol, but the binding energy of the mutant protein is −5.97 kcal/mol. Replacing the active site with amino acids other than alanine had no effect on the activity of either asparaginase or glutaminase. As a result, an enzyme model with reduced glutaminase activity was created [87].
The effect of the N248S mutation on L-ASNase activity has been studied by Aghaeepoor et al. Molecular dynamics simulations, molecular docking and quantum mechanical/molecular mechanics studies are just some of the in silico analyses that have been carried out. The AutoDock Vina program was used for molecular docking of the substrates asparagine and glutamine in the active site of mutant L-ASNase. The N248S mutation was associated with strong L-ASNase activity, whereas glutaminase activity was decreased, according to the in silico analysis and experimental evidence [88].
4.3. Modifications to Increase Catalytic Activity and Specificity
An in silico method was developed to predict the kinetics (Km value) of L-ASNase from an enzyme sequence and to test for the optimal alternative L-ASNase II against ALL. The E. coli asnB sequence was utilized to look for homologous proteins in different bacterial and archaeal phyla. This method uses sequence-based phylogenetic analysis to narrow down a small number of candidates on which virtual docking can be utilized to identify a set of optimal enzymes that may outperform commercially available enzymes. Mega-X software (https://www.megasoftware.net/) was used to create a maximum likelihood phylogenetic tree. In terms of immunogenicity, the sequences most distant from E. coli and Erwinia species were regarded as the best candidates and were chosen for further processing. The architectures of these proteins were created using homology modeling in MODELLER 9.22. Following model validation, PyMol was used to discover active sites for each protein, and asparagine was docked with these proteins to compute binding (AutoDock Vina coupled with PyRxsoftware). As a result, three potentially promising L-ASNase II enzymes produced by three Streptomyces species were predicted. They have the highest binding energy (−5.3 kcal/mol, −5.2 kcal/mol, and −5.3 kcal/mol, respectively; greater than E. coli and Erwinia L-ASNase) and were predicted to have the lowest Kms since binding energy and Km have an inverse relationship [89].
Rational modification of L-ASNase from Bacillus licheniformis (BliA) was carried out to obtain an enzyme with better catalytic properties and half-life. This L-ASNase was chosen because natural BliA lacks glutaminase activity. MSA of L-ASNase protein sequences from E. coli (EcA), Erwinia carotovora (ErA), Pseudomonas sp. (PGA), P. furiosus (PfA) and B. licheniformis using CLUSTAL 2.1 showed that BliA is very similar to PfA compared to EcA, ErA and PGA. BliA was altered by site-directed mutagenesis using the overlap extension approach. Based on the sequence alignment of previously mutagenized L-ASNases, D103 and G238, four alternative sites, D103, G238, E232 and Q112, were selected to improve hydrophobicity at the dimer interface and increase electrostatic potential. A methodical approach to enzyme engineering resulted in four mutants: G238N, E232A, D103V and Q112H. The mutant D103V enzyme had a specific activity of 597.7 IU/mg, which was higher than that of the native (rBliA) enzyme (407.65 IU/mg). In addition, the D103V mutant BliA was superior to natural BliA at the optimal temperature and in vitro half-life, showing resistance to higher temperatures and a 3-fold longer half-life. Kinetic experiments showed that the D103V mutant L-ASNase has a higher substrate affinity, with a Km of 0.42 mM and a Vmax of 2778.9 µmol min−1. Thus, the change from D to V at position 103 is responsible for the improved catalytic properties of BliA [90].
4.4. Modifications to Increase Thermostability
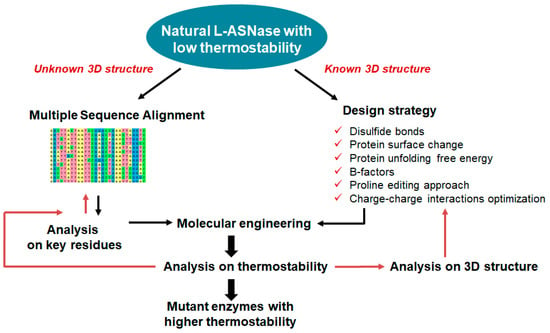
The improvement in the thermostability of L-ASNase is crucial for the extension of the range of applications of L-ASNase in the preprocessing of food [91]. The thermal stability of L-ASNase can effectively enhance its ability to reduce the amount of acrylamide [92]. To prevent the formation of acrylamide, different forms of L-ASNase can be used in fried potato products [93]. Random mutagenesis and B-factor screening are often used to improve protein thermostability at the expense of screening thousands of clones (see Figure 6). The computational design of stability-enhancing mutations is a reliable alternative to screening-based approaches, but reliable methods need to be developed to improve prediction accuracy [94]. Several studies have used rational design to develop L-ASNases with increased thermal stability.
Figure 6. Rational design strategies for improving the thermostability of L-ASNase. Rational design can be used to identify structural flaws in proteins and modify them to increase thermostability: (1) in the case of unknown 3D structure (left), information was obtained by sequence analysis with construct MSA. The most abundant amino acid at each position in a protein sequence often significantly affects stability. Then, 3D modeling, protein stability prediction algorithms and others are used to analyze key residues and predicted residue contacts; (2) when the 3D structure of a protein is known (right), it can be examined to find regions that are prone to unfolding or destabilization at high temperatures. The overall thermostability of the protein can be improved by strengthening these regions using molecular dynamics, strategies based on disulfide bonds, protein surface charge optimization, homologous comparison strategies, design strategy using proline effect analysis and genetic engineering.
Jiao et al. used heterologic expression in E. coli BL21(DE3) and consensus design in the Consensus Finder web server to mutate cysteine residues to improve the thermostability of L-ASNase from Acinetobacter soli Y-3 (AsA). AcA has low thermal stability due to unstable cysteine residues and mismatched intermolecular disulfide bonds. Cys8 and Cys283 of Acinetobacter soli L-ASNase (AsA) were screened for non-conserved cysteine residues with readily oxidizable free sulfhydryl groups, and mutagenesis was performed. A C8Y/C283Q mutant with dramatically increased thermal stability was produced by saturation mutagenesis and combinatorial mutation, with a half-life of 361.6 min at 40 °C, more than 34 times longer than that of the wild type. Its melting point (Tm) is 62.3 degrees Celsius, which is 7.1 degrees Celsius higher than that of the wild type. The evolution of novel hydrogen bonds of Gln283 and the aromatic interaction of Tyr8 with neighboring residues were identified using molecular dynamics modeling in GROMACS and structural analysis in the SWISS-MODEL online server. Thus, the additional hydrogen bonds and conjugation forces induced by the mutation contribute significantly to the heat stability of the mutants. The acrylamide concentration of potato chips was efficiently reduced by 86.50% after treatment with the C8Y/C283Q mutant, which was much higher than the 59.05% reduction after treatment with wild-type AsA [92].
The thermal stability of BlAsnase from Bacillus licheniformis was improved using rational design methods. PoPMuSiC was used to predict important possible mutation hotspots and appropriate replacements to increase the thermal stability of the L-ASNase protein. Ranking was used to select amino acid regions with low free energy values based on folding free energy calculations. Furthermore, their conservation was checked by sequence comparison, and the spatial position of the corresponding amino acids in the protein structure was observed using PyMOL to avoid the negative effect of mutations at specific sites on the enzymatic activity of BlAsnase, thus eliminating some inappropriate mutation sites. AMBER 18 software was used to perform all-atom molecular dynamics simulations on dimeric compounds. The structure is rigid, there are hydrophobic contacts, and the electrostatic potential is favorable. The results showed that the D172 W/E207A double mutant had a much stiffer and more stable protein structure than the wild-type BlAsnase. This mutant outperformed wild-type BlAsnase in terms of thermal stability, with a 65.8-fold longer half-life at 55 °C and a 5 °C higher optimum reaction temperature and melting point [95].
4.5. Modifications to Decrease Immunogenicity
Protein immunogenicity can be reduced by altering regions within the target protein that are likely to be recognized as B- or T-cell epitopes by the adaptive immune system. However, the identification and removal of B-cell epitopes is extremely challenging due to their conformational nature. This is further complicated by a poor understanding of the naive antibody repertoire and how it changes in different human populations. Mutagenesis has been successfully used to reduce the immunogenicity of humanized and chimeric antibodies by removing T-cell epitopes. However, although these proteins have only a few relatively short potentially immunogenic regions, heterologous enzymes that have not been immunologically tolerated are likely to have many T-cell epitopes. Extensive mutagenesis of the polypeptide sequence, while retaining the original catalytic activity, would therefore be required to remove these epitopes. Furthermore, in addition to the active site residues, a network of amino acids present throughout the protein influences enzyme catalysis [96].
Computational techniques to address protein immunogenicity issues are gaining popularity. Deimmunogenization of therapeutic proteins by altering T-cell or B-cell epitopes using bioinformatic tools and site-directed mutagenesis has been shown to be an effective technique for creating safer biopharmaceuticals. Based on these concepts and the high immunogenicity of E. coli L-ASNase in 2019, Belén et al. created an in silico chimera of L-ASNase by replacing epitope peptides in the E. coli enzyme variant with peptides from human serum albumin and even demonstrated proof of concept that the engineered variant is recombinantly expressed in E. coli [97].
5. Conclusions
The evolution of host strains has been influenced by changes at many different levels, including site-directed mutagenesis, promoters, transcription factors, gene copy number, codons, chaperones, leader peptides and structural levels such as glycosylation and enzyme folding [98].
Through improving the physicochemical properties of engineered proteins, selecting the best cell line, and optimizing culture media and expression conditions, it is possible to produce recombinant proteins more efficiently and with higher yields while reducing the number of time-consuming and costly steps involved. Experimental methods for screening potential drug compounds are still very expensive and take a long time. A more efficient method is to pretest a large library of small molecules in silico and then select a small group for experimental validation. In addition, the availability of data on the three-dimensional structure of enzymes and substrates makes it possible to analyze their interaction energy and other parameters [89]. Computational approaches are often used in pharmaceutical biotechnology research to create new types of protein therapeutics with improved performance [99].
Today, the application of modern computational tools and directed evolution approaches can alter the evolution of enzymes. The combination of directed evolution with rational and semi-rational strategies, including molecular dynamics and quantum mechanics/molecular mechanics simulations, is expected to lead to the development of enzymes with improved biotechnological properties in the coming years. It is a promising approach to identify targets for protein engineering and reduce library screening efforts [100]. Therefore, suitable heterologous expression systems with tolerance to high enzyme expression levels should be developed for enzymes. For example, directed evolution combined with hybrid techniques has led to functional heterologous expression and activity, adaptation to non-natural habitats and the creation of laccase chimeras with mixed characteristics [101]. Rational design and heterologous expression in E. coli were used to obtain a new version of the thrombolytic drug, Reteplase [102].
Recently, the thermostability of L-ASNase from R. miehei and Acinetobacter soli has been improved via heterologous expression and rational design [92][103]. This approach also resulted in L-ASNase with improved stability, half-life, reduced immunogenicity, in vitro and in vivo anti-cancer activity. In addition, this L-ASNase was more active than the native or PEG-conjugated enzyme [104].
Therefore, in silico modeling has recently emerged as a critical technique for identifying defects at the molecular level, and the data collected can be used to modify the native L-ASNase structure. Because this method avoids costly, labor-intensive and time-consuming experimental designs, it may become more common in the future [105]. Rational design can provide insight into the structure-function relationship of L-ASNase. By studying the effects of specific mutations on the activity and stability of the enzyme, researchers can gain a better understanding of the structure and function of the protein. This knowledge can be used to guide future protein engineering efforts and improve the efficiency of the rational design process.
References
- Pokrovskaya, M.V.; Pokrovsky, V.S.; Aleksandrova, S.S.; Sokolov, N.N.; Zhdanov, D.D. Molecular Analysis of L-Asparaginases for Clarification of the Mechanism of Action and Optimization of Pharmacological Functions. Pharmaceutics 2022, 14, 599.
- Sankaran, H.; Sengupta, S.; Purohit, V.; Kotagere, A.; Moulik, N.R.; Prasad, M.; Dhamne, C.; Narula, G.; Banavali, S.; Gota, V. A Comparison of Asparaginase Activity in Generic Formulations of E. coli Derived L-Asparaginase: In-vitro Study and Retrospective Analysis of Asparaginase Monitoring in Pediatric Patients with Leukemia. Br. J. Clin. Pharmacol. 2020, 86, 1081–1088.
- Ferguson, D.C.; McCorkle, J.R.; Barnett, K.R.; Bonten, E.J.; Bergeron, B.P.; Bhattarai, K.R.; Yang, W.; Smith, C.; Hansen, B.S.; Bajpai, R.; et al. Amino Acid Stress Response Genes Promote L-Asparaginase Resistance in Pediatric Acute Lymphoblastic leukemia. Blood Adv. 2022, 6, 3386–3397.
- Vimal, A.; Kumar, A. L-Asparaginase: A Feasible Therapeutic Molecule for Multiple Diseases. 3 Biotech 2018, 8, 278.
- Völler, S.; Pichlmeier, U.; Zens, A.; Hempel, G. Pharmacokinetics of Recombinant Asparaginase in Children with Acute Lymphoblastic leukemia. Cancer Chemother. Pharmacol. 2018, 81, 305–314.
- Van Trimpont, M.; Peeters, E.; De Visser, Y.; Schalk, A.M.; Mondelaers, V.; De Moerloose, B.; Lavie, A.; Lammens, T.; Goossens, S.; Van Vlierberghe, P. Novel Insights on the Use of L-Asparaginase as an Efficient and Safe Anti-Cancer Therapy. Cancers 2022, 14, 902.
- Mazloum-Ravasan, S.; Madadi, E.; Mohammadi, A.; Mansoori, B.; Amini, M.; Mokhtarzadeh, A.; Baradaran, B.; Darvishi, F. Yarrowia Lipolytica L-Asparaginase Inhibits the Growth and Migration of Lung (A549) and Breast (MCF7) Cancer Cells. Int. J. Biol. Macromol. 2021, 170, 406–414.
- Darvishi, F.; Jahanafrooz, Z.; Mokhtarzadeh, A. Microbial L-Asparaginase as a Promising Enzyme for Treatment of Various Cancers. Appl. Microbiol. Biotechnol. 2022, 106, 5335–5347.
- Ravi, A.; Gurunathan, B. Acrylamide Mitigation in Fried Kochchi Kesel Chips Using Free and Immobilized Fungal Asparaginase. Food Technol. Biotechnol. 2018, 56, 51–57.
- Gazi, S.; Göncüoğlu Taş, N.; Görgülü, A.; Gökmen, V. Effectiveness of Asparaginase on Reducing Acrylamide Formation in Bakery Products according to Their Dough Type and Properties. Food Chem. 2023, 402, 134224.
- Sajed, M.; Ahmad, N.; Rashid, N. Temperature Dependent Autocleavage and Applications of Recombinant L-Asparaginase from Thermococcus Kodakarensis for Acrylamide Mitigation. 3 Biotech 2022, 12, 129.
- Bosmann, H.B.; Kessel, D. Inhibition of Glycoprotein Synthesis in L5178Y Mouse Lukaemic Cells by L-Asparaginase in Vitro. Nature 1970, 226, 850–851.
- Plyasova, A.A.; Pokrovskaya, M.V.; Lisitsyna, O.M.; Pokrovsky, V.S.; Alexandrova, S.S.; Hilal, A.; Sokolov, N.N.; Zhdanov, D.D. Penetration into Cancer Cells via Clathrin-dependent Mechanism Allows L-asparaginase from Rhodospirillum rubrum to Inhibit Telomerase. Pharmaceuticals 2020, 13, 286.
- Ankel, E.G.; Zirneski, J.; Ring, B.J.; Holcenberg, J.S. Effect of Asparaginase on Cell Membranes of Sensitive and Resistants Mouse Lymphoma Cells. Vitr. Cell. Dev. Biol.-Plant 1984, 20, 376–384.
- Brumano, L.P.; da Silva, F.V.S.; Costa-Silva, T.A.; Apolinário, A.C.; Santos, J.H.P.M.; Kleingesinds, E.K.; Monteiro, G.; Rangel-Yagui, C.d.O.; Benyahia, B.; Junior, A.P. Development of L-Asparaginase Biobetters: Current Research Status and Review of the Desirable Quality Profiles. Front. Bioeng. Biotechnol. 2018, 6, 212.
- da Cunha, M.C.; dos Santos Aguilar, J.G.; de Melo, R.R.; Nagamatsu, S.T.; Ali, F.; de Castro, R.J.S.; Sato, H.H. Fungal L-Asparaginase: Strategies for Production and Food Applications. Food Res. Int. 2019, 126, 108658.
- Wang, Y.; Xu, W.; Wu, H.; Zhang, W.; Guang, C.; Mu, W. Microbial Production, Molecular Modification, and Practical Application of l-Asparaginase: A Review. Int. J. Biol. Macromol. 2021, 186, 975–983.
- Chakravarty, N.; Priyanka; Singh, J.; Singh, R.P. A Potential Type-II L-Asparaginase from Marine Isolate Bacillus australimaris NJB19: Statistical Optimization, in Silico Analysis and Structural Modeling. Int. J. Biol. Macromol. 2021, 174, 527–539.
- Mahajan, R.V.; Kumar, V.; Rajendran, V.; Saran, S.; Ghosh, P.C.; Saxena, R.K. Purification and Characterization of a Novel and Robust L-Asparaginase Having Low-Glutaminase Activity from Bacillus licheniformis: In Vitro Evaluation of Anti-Cancerous Properties. PLoS ONE 2014, 9, e99037.
- Cachumba, J.J.M.; Antunes, F.A.F.; Peres, G.F.D.; Brumano, L.P.; Dos Santos, J.C.; Da Silva, S.S. Current Applications and Different Approaches for Microbial L-Asparaginase Production. Braz. J. Microbiol. Publ. Braz. Soc. Microbiol. 2016, 47 (Suppl. S1), 77–85.
- Rieder, L.; Teuschler, N.; Ebner, K.; Glieder, A. Eukaryotic Expression Systems for Industrial Enzymes. In Industrial Enzyme Applications; Wiley: Hoboken, NJ, USA, 2019; pp. 47–69.
- Al-Hazmi, N.E.; Naguib, D.M. Plant Asparaginase versus Microbial Asparaginase as Anticancer Agent. Environ. Sci. Pollut. Res. 2022, 29, 27283–27293.
- Lopes, A.M.; de Oliveira-Nascimento, L.; Ribeiro, A.; Tairum, C.A.J.; Breyer, C.A.; de Oliveira, M.A.; Monteiro, G.; de Souza-Motta, C.M.; Magalhães, P.d.O.; Avendaño, J.G.F.; et al. Therapeutic L-Asparaginase: Upstream, Downstream and Beyond. Crit. Rev. Biotechnol. 2017, 37, 82–99.
- Darvishi, F.; Faraji, N.; Shamsi, F. Production and Structural Modeling of a Novel Asparaginase in Yarrowia lipolytica. Int. J. Biol. Macromol. 2019, 125, 955–961.
- Chand, S.; Mahajan, R.V.; Prasad, J.P.; Sahoo, D.K.; Mihooliya, K.N.; Dhar, M.S.; Sharma, G. A Comprehensive Review on Microbial L-Asparaginase: Bioprocessing, Characterization, and Industrial Applications. Biotechnol. Appl. Biochem. 2020, 67, 619–647.
- Orabi, H.M.; El-Fakharany, E.M.; Abdelkhalek, E.S.; Sidkey, N.M. L-Asparaginase And L-Glutaminase: Sources, Production, and Applications In Medicine and Industry. J. Microbiol. Biotechnol. Food Sci. 2019, 9, 179–190.
- Dantas, R.C.; Caetano, L.F.; Torres, A.L.S.; Alves, M.S.; Silva, E.T.M.F.; Teixeira, L.P.R.; Teixeira, D.C.; de Azevedo Moreira, R.; Fonseca, M.H.G.; Gaudêncio Neto, S.; et al. Expression of a Recombinant Bacterial L-Asparaginase in Human Cells. BMC Res. Notes 2019, 12, 794.
- L-Asparaginase (ASRGL1) Recombinant Protein. Available online: https://www.mybiosource.com/recombinant-protein/l-asparaginase-asrgl1/1420071 (accessed on 11 October 2023).
- Castro, D.; Marques, A.S.C.; Almeida, M.R.; de Paiva, G.B.; Bento, H.B.S.; Pedrolli, D.B.; Freire, M.G.; Tavares, A.P.M.; Santos-Ebinuma, V.C. L-Asparaginase Production Review: Bioprocess Design and Biochemical Characteristics. Appl. Microbiol. Biotechnol. 2021, 105, 4515–4534.
- Izadpanah Qeshmi, F.; Homaei, A.; Fernandes, P.; Javadpour, S. Marine Microbial L-Asparaginase: Biochemistry, Molecular Approaches and Applications in Tumor Therapy and in Food Industry. Microbiol. Res. 2018, 208, 99–112.
- Loch, J.I.; Jaskolski, M. Structural and Biophysical Aspects of L-Asparaginases: A Growing Family with Amazing Diversity. IUCrJ 2021, 8, 514–531.
- Michalska, K.; Jaskolski, M. Structural Aspects of L-Asparaginases, Their Friends and Relations. Acta Biochim. Pol. 2006, 53, 627–640.
- Bejger, M.; Imiolczyk, B.; Clavel, D.; Gilski, M.; Pajak, A.; Marsolais, F.; Jaskolski, M. Na+/K+ Exchange Switches the Catalytic Apparatus of Potassium-Dependent Plant L-Asparaginase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 1854–1872.
- Dumina, M.; Zhgun, A. Thermo-L-Asparaginases: From the Role in the Viability of Thermophiles and Hyperthermophiles at High Temperatures to a Molecular Understanding of Their Thermoactivity and Thermostability. Int. J. Mol. Sci. 2023, 24, 2674.
- Lubkowski, J.; Wlodawer, A. Geometric Considerations Support the Double-displacement Catalytic Mechanism of l-asparaginase. Protein Sci. 2019, 28, 1850–1864.
- Kumar, S.; Darnal, S.; Patial, V.; Kumar, V.; Singh, D. Molecular Characterization of a Stable and Robust L-Asparaginase from Pseudomonas sp. PCH199: Evaluation of Cytotoxicity and Acrylamide Mitigation Potential. Fermentation 2022, 8, 568.
- Gesto, D.S.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Unraveling the Enigmatic Mechanism of L-Asparaginase II with QM/QM Calculations. J. Am. Chem. Soc. 2013, 135, 7146–7158.
- Fonseca, M.H.G.; Fiúza, T.d.S.; de Morais, S.B.; de Souza, T.d.A.C.B.; Trevizani, R. Circumventing the Side Effects of L-Asparaginase. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 139, 111616.
- Aghaiypour, K.; Wlodawer, A.; Lubkowski, J. Structural Basis for the Activity and Substrate Specificity of Erwinia chrysanthemi L-Asparaginase. Biochemistry 2001, 40, 5655–5664.
- Jaskólski, M.; Kozak, M.; Lubkowski, J.; Palm, G.; Wlodawer, A. Structures of Two Highly Homologous Bacterial L-Asparaginases: A Case of Enantiomorphic Space Groups. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 369–377.
- Gupta, S.K.; Shukla, P. Advanced Technologies for Improved Expression of Recombinant Proteins in Bacteria: Perspectives and Applications. Crit. Rev. Biotechnol. 2016, 36, 1089–1098.
- Asitok, A.; Ekpenyong, M.; Amenaghawon, A.; Akwagiobe, E.; Asuquo, M.; Rao, A.; Ubi, D.; Iheanacho, J.; Etiosa, J.; Antai, A.; et al. Production, Characterization and Techno-Economic Evaluation of Aspergillus Fusant l-Asparaginase. AMB Express 2023, 13, 2.
- Lawson, C.E.; Martí, J.M.; Radivojevic, T.; Jonnalagadda, S.V.R.; Gentz, R.; Hillson, N.J.; Peisert, S.; Kim, J.; Simmons, B.A.; Petzold, C.J.; et al. Machine Learning for Metabolic Engineering: A Review. Metab. Eng. 2021, 63, 34–60.
- Shakambari, G.; Sameer Kumar, R.; Ashokkumar, B.; Ganesh, V.; Vasantha, V.S.; Varalakshmi, P. Cloning and Expression of L-Asparaginase from Bacillus tequilensis PV9W and Therapeutic Efficacy of Solid Lipid Particle Formulations against Cancer. Sci. Rep. 2018, 8, 18013.
- Kim, S.-K.; Min, W.-K.; Park, Y.-C.; Seo, J.-H. Application of Repeated Aspartate Tags to Improving Extracellular Production of Escherichia coli L-Asparaginase Isozyme II. Enzym. Microb. Technol. 2015, 79–80, 49–54.
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172.
- Baeshen, N.A.; Baeshen, M.N.; Sheikh, A.; Bora, R.S.; Ahmed, M.M.M.; Ramadan, H.A.I.; Saini, K.S.; Redwan, E.M. Cell Factories for Insulin Production. Microb. Cell Factories 2014, 13, 141.
- Plomp, P.J.A.M.; Boer, L.D.; Rooijen, R.J.V.; Meima, R.B. Asparaginase and Its Use in Food Production. Available online: https://patents.google.com/patent/US8105815B2/en (accessed on 11 October 2023).
- Pouresmaeil, M.; Azizi-Dargahlou, S. Factors Involved in Heterologous Expression of Proteins in E. coli Host. Arch. Microbiol. 2023, 205, 212.
- Heyde, S.A.H.; Nørholm, M.H.H. Tailoring the Evolution of BL21(DE3) Uncovers a Key Role for RNA Stability in Gene Expression Toxicity. Commun. Biol. 2021, 4, 963.
- Radha, R.; Arumugam, N.; Gummadi, S.N. Glutaminase Free L-Asparaginase from Vibrio Cholerae: Heterologous Expression, Purification and Biochemical Characterization. Int. J. Biol. Macromol. 2018, 111, 129–138.
- Einsfeldt, K.; Baptista, I.C.; Pereira, J.C.C.V.; Costa-Amaral, I.C.; da Costa, E.S.; Ribeiro, M.C.M.; Land, M.G.P.; Alves, T.L.M.; Larentis, A.L.; Almeida, R.V. Recombinant L-Asparaginase from Zymomonas Mobilis: A Potential New Antileukemic Agent Produced in Escherichia coli. PLoS ONE 2016, 11, e0156692.
- Studier, F.W.; Daegelen, P.; Lenski, R.E.; Maslov, S.; Kim, J.F. Understanding the Differences between Genome Sequences of Escherichia coli B Strains REL606 and BL21(DE3) and Comparison of the E. coli B and K-12 Genomes. J. Mol. Biol. 2009, 394, 653–680.
- Pourhossein, M.; Korbekandi, H. Cloning, Expression, Purification and Characterisation of Erwinia Carotovora L-Asparaginase in Escherichia coli. Adv. Biomed. Res. 2014, 3, 82.
- de Moura, W.A.F.; Schultz, L.; Breyer, C.A.; de Oliveira, A.L.P.; Tairum, C.A.; Fernandes, G.C.; Toyama, M.H.; Pessoa, A., Jr.; Monteiro, G.; de Oliveira, M.A. Functional and Structural Evaluation of the Antileukaemic Enzyme L-Asparaginase II Expressed at Low Temperature by Different Escherichia coli Strains. Biotechnol. Lett. 2020, 42, 2333–2344.
- Gomes, A.R.; Byregowda, S.M.; Veeregowda, B.M.; Balamurugan, V. An Overview of Heterologous Expression Host Systems for the Production of Recombinant Proteins. Adv. Anim. Vet. Sci. 2016, 4, 346–356.
- de Souza, C.C.; Guimarães, J.M.; Pereira, S.d.S.; Mariúba, L.A.M. The Multifunctionality of Expression Systems in Bacillus subtilis: Emerging Devices for the Production of Recombinant Proteins. Exp. Biol. Med. 2021, 246, 2443–2453.
- Yang, H.; Qu, J.; Zou, W.; Shen, W.; Chen, X. An Overview and Future Prospects of Recombinant Protein Production in Bacillus subtilis. Appl. Microbiol. Biotechnol. 2021, 105, 6607–6626.
- Cui, W.; Han, L.; Suo, F.; Liu, Z.; Zhou, L.; Zhou, Z. Exploitation of Bacillus subtilis as a Robust Workhorse for Production of Heterologous Proteins and Beyond. World J. Microbiol. Biotechnol. 2018, 34, 145.
- Zhao, X.; Xu, J.; Tan, M.; Zhen, J.; Shu, W.; Yang, S.; Ma, Y.; Zheng, H.; Song, H. High Copy Number and Highly Stable Escherichia coli–Bacillus subtilis Shuttle Plasmids Based on PWB980. Microb. Cell Factories 2020, 19, 25.
- Nguyen, H.D.; Nguyen, Q.A.; Ferreira, R.C.; Ferreira, L.C.S.; Tran, L.T.; Schumann, W. Construction of Plasmid-Based Expression Vectors for Bacillus subtilis Exhibiting Full Structural Stability. Plasmid 2005, 54, 241–248.
- Schumann, W. Production of Recombinant Proteins in Bacillus subtilis. Adv. Appl. Microbiol. 2007, 62, 137–189.
- Krüger, A.; Welsch, N.; Dürwald, A.; Brundiek, H.; Wardenga, R.; Piascheck, H.; Mengers, H.G.; Krabbe, J.; Beyer, S.; Kabisch, J.F.; et al. A Host-Vector Toolbox for Improved Secretory Protein Overproduction in Bacillus subtilis. Appl. Microbiol. Biotechnol. 2022, 106, 5137–5151.
- Pan, Y.; Yang, J.; Wu, J.; Yang, L.; Fang, H. Current Advances of Pichia pastoris as Cell Factories for Production of Recombinant Proteins. Front. Microbiol. 2022, 13, 1059777.
- Daly, R.; Hearn, M.T.W. Expression of Heterologous Proteins in Pichia pastoris: A Useful Experimental Tool in Protein Engineering and Production. J. Mol. Recognit. 2005, 18, 119–138.
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A Highly Successful Expression System for Optimal Synthesis of Heterologous Proteins. J. Cell. Physiol. 2020, 235, 5867–5881.
- Mohammadzadeh, R.; Karbalaei, M.; Soleimanpour, S.; Mosavat, A.; Rezaee, S.A.; Ghazvini, K.; Farsiani, H. Practical Methods for Expression of Recombinant Protein in the Pichia pastoris System. Curr. Protoc. 2021, 1, e155.
- Macauley-Patrick, S.; Fazenda, M.L.; McNeil, B.; Harvey, L.M. Heterologous Protein Production Using the Pichia pastoris Expression System. Yeast 2005, 22, 249–270.
- Li, P.; Anumanthan, A.; Gao, X.-G.; Ilangovan, K.; Suzara, V.V.; Düzgüneş, N.; Renugopalakrishnan, V. Expression of Recombinant Proteins in Pichia pastoris. Appl. Biochem. Biotechnol. 2007, 142, 105–124.
- Juturu, V.; Wu, J.C. Heterologous Protein Expression in Pichia pastoris: Latest Research Progress and Applications. Chembiochem A Eur. J. Chem. Biol. 2018, 19, 7–21.
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein Expression in Pichia pastoris: Recent Achievements and Perspectives for Heterologous Protein Production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317.
- Safder, I.; Islam, I.-U.; Kazim, M.; Khan, S. Pichia pastoris Expression System: A Potential Candidate to Express Protein in Industrial and Biopharmaceutical Domains. Biomed. Lett. 2018, 4, 1–14.
- Gao, J.; Jiang, L.; Lian, J. Development of Synthetic Biology Tools to Engineer Pichia pastoris as a Chassis for the Production of Natural Products. Synth. Syst. Biotechnol. 2021, 6, 110–119.
- Thor, D.; Xiong, S.; Orazem, C.; Kwan, A.; Cregg, J.; Lincereghino, J.; Lincereghino, G. Cloning and Characterization of the Gene as a Selectable Marker. FEMS Yeast Res. 2005, 5, 935–942.
- Piva, L.C.; Bentacur, M.O.; Reis, V.C.B.; De Marco, J.L.; de Moraes, L.M.P.; Torres, F.A.G. Molecular Strategies to Increase the Levels of Heterologous Transcripts in Komagataella Phaffii for Protein Production. Bioengineered 2017, 8, 441–445.
- Erden-Karaoğlan, F.; Karaoğlan, M. Improvement of Recombinant L-Asparaginase Production in Pichia pastoris. 3 Biotech 2023, 13, 164.
- Song, Z.; Zhang, Q.; Wu, W.; Pu, Z.; Yu, H. Rational Design of Enzyme Activity and Enantioselectivity. Front. Bioeng. Biotechnol. 2023, 11, 1129149.
- Korendovych, I.V. Rational and Semirational Protein Design. Methods Mol. Biol. 2018, 1685, 15–23.
- Yang, G.; Miton, C.M.; Tokuriki, N. A Mechanistic View of Enzyme Evolution. Protein Sci. 2020, 29, 1724–1747.
- Mortazavi, M.; Torkzadeh-Mahani, M.; Kargar, F.; Nezafat, N.; Ghasemi, Y. In Silico Analysis of Codon Usage and Rare Codon Clusters in the Halophilic Bacteria L-Asparaginase. Biologia 2020, 75, 151–160.
- Orhan, H.; Aktaş Uygun, D. Immobilization of L-Asparaginase on Magnetic Nanoparticles for Cancer Treatment. Appl. Biochem. Biotechnol. 2020, 191, 1432–1443.
- Kante, R.K.; Somavarapu, S.; Vemula, S.; Kethineni, C.; Mallu, M.R.; Ronda, S.R. Production of Recombinant Human Asparaginase from Escherichia coli under Optimized Fermentation Conditions: Effect of Physicochemical Properties on Enzyme Activity. Biotechnol. Bioprocess Eng. 2019, 24, 824–832.
- González-Torres, I.; Perez-Rueda, E.; Evangelista-Martínez, Z.; Zárate-Romero, A.; Moreno-Enríquez, A.; Huerta-Saquero, A. Identification of L-Asparaginases from Streptomyces Strains with Competitive Activity and Immunogenic Profiles: A Bioinformatic Approach. PeerJ 2020, 8, e10276.
- Nguyen, H.A.; Su, Y.; Lavie, A. Structural Insight into Substrate Selectivity of Erwinia Chrysanthemi L-Asparaginase. Biochemistry 2016, 55, 1246–1253.
- Nguyen, H.A.; Su, Y.; Lavie, A. Design and Characterization of Erwinia Chrysanthemi L-Asparaginase Variants with Diminished l-Glutaminase Activity. J. Biol. Chem. 2016, 291, 17664–17676.
- Chan, W.K.; Lorenzi, P.L.; Anishkin, A.; Purwaha, P.; Rogers, D.M.; Sukharev, S.; Rempe, S.B.; Weinstein, J.N. The Glutaminase Activity of L-Asparaginase Is Not Required for Anticancer Activity against ASNS-Negative Cells. Blood 2014, 123, 3596–3606.
- LN, R.; Doble, M.; Rekha, V.P.B.; Pulicherla, K.K. In Silico Engineering of L-Asparaginase to Have Reduced Glutaminase Side Activity for Effective Treatment of Acute Lymphoblastic leukemia. J. Pediatr. Hematol./Oncol. 2011, 33, 617–621.
- Aghaeepoor, M.; Akbarzadeh, A.; Mirzaie, S.; Hadian, A.; Jamshidi Aval, S.; Dehnavi, E. Selective Reduction in Glutaminase Activity of L-Asparaginase by Asparagine 248 to Serine Mutation: A Combined Computational and Experimental Effort in Blood Cancer Treatment. Int. J. Biol. Macromol. 2018, 120, 2448–2457.
- Baral, A.; Gorkhali, R.; Basnet, A.; Koirala, S.; Bhattarai, H.K. Selection of the Optimal L-Asparaginase II Against Acute Lymphoblastic leukemia: An In Silico Approach. JMIRx Med. 2021, 2, e29844.
- Sudhir, A.P.; Agarwaal, V.V.; Dave, B.R.; Patel, D.H.; Subramanian, R.B.B. Enhanced Catalysis of L-Asparaginase from Bacillus licheniformis by a Rational Redesign. Enzym. Microb. Technol. 2016, 86, 1–6.
- Zhang, W.; Dai, Q.; Huang, Z.; Xu, W. Identification and Thermostability Modification of the Mesophilic L-Asparaginase from Limosilactobacillus secaliphilus. Appl. Biochem. Biotechnol. 2023, 1–15.
- Jiao, L.; Chi, H.; Xia, B.; Lu, Z.; Bie, X.; Zhao, H.; Lu, F.; Chen, M. Thermostability Improvement of L-Asparaginase from Acinetobacter Soli via Consensus-Designed Cysteine Residue Substitution. Molecules 2022, 27, 6670.
- Jia, R.; Wan, X.; Geng, X.; Xue, D.; Xie, Z.; Chen, C. Microbial L-Asparaginase for Application in Acrylamide Mitigation from Food: Current Research Status and Future Perspectives. Microorganisms 2021, 9, 1659.
- Li, G.; Fang, X.; Su, F.; Chen, Y.; Xu, L.; Yan, Y. Enhancing the Thermostability of Rhizomucor miehei Lipase with a Limited Screening Library by Rational-Design Point Mutations and Disulfide Bonds. Appl. Environ. Microbiol. 2018, 84, e02129-17.
- Chi, H.; Wang, Y.; Xia, B.; Zhou, Y.; Lu, Z.; Lu, F.; Zhu, P. Enhanced Thermostability and Molecular Insights for L -Asparaginase from Bacillus licheniformis Structure- and Computation-Based Rational Design. J. Agric. Food Chem. 2022, 70, 14499–14509.
- Cantor, J.R.; Panayiotou, V.; Agnello, G.; Georgiou, G.; Stone, E.M. Engineering Reduced-Immunogenicity Enzymes for Amino Acid Depletion Therapy in Cancer. Methods Enzymol. 2012, 502, 291–319.
- Belén, L.H.; Lissabet, J.B.; de Oliveira Rangel-Yagui, C.; Effer, B.; Monteiro, G.; Pessoa, A.; Farías Avendaño, J.G. A Structural in Silico Analysis of the Immunogenicity of L-Asparaginase from Escherichia coli and Erwinia carotovora. Biologicals 2019, 59, 47–55.
- Dadwal, A.; Sharma, S.; Satyanarayana, T. Progress in Ameliorating Beneficial Characteristics of Microbial Cellulases by Genetic Engineering Approaches for Cellulose saccharification. Front. Microbiol. 2020, 11, 1387.
- Sinha, R.; Shukla, P. Current Trends in Protein Engineering: Updates and Progress. Curr. Protein Pept. Sci. 2019, 20, 398–407.
- Pardo, I.; Camarero, S. Laccase Engineering by Rational and Evolutionary Design. Cell. Mol. Life Sci. 2015, 72, 897–910.
- Mate, D.M.; Alcalde, M. Laccase Engineering: From Rational Design to Directed Evolution. Biotechnol. Adv. 2015, 33, 25–40.
- Seyedhosseini Ghaheh, H.; Sajjadi, S.; Shafiee, F.; Barzegari, E.; Moazen, F.; Mir Mohammad Sadeghi, H. Rational Design of a New Variant of Reteplase with Optimized Physicochemical Profile and Large-Scale Production in Escherichia coli. World J. Microbiol. Biotechnol. 2022, 38, 29.
- Zhang, X.; Wang, Z.; Wang, Y.; Li, X.; Zhu, M.; Zhang, H.; Xu, M.; Yang, T.; Rao, Z. Heterologous Expression and Rational Design of L-Asparaginase from Rhizomucor miehei to Improve Thermostability. Biology 2021, 10, 1346.
- Zhang, S.; Sun, Y.; Zhang, L.; Zhang, F.; Gao, W. Thermoresponsive polypeptide Fused L-Asparaginase with Mitigated Immunogenicity and Enhanced Efficacy in Treating Hematologic malignancies. Adv. Sci. 2023, 10, e2300469.
- Patel, P.G.; Panseriya, H.Z.; Vala, A.K.; Dave, B.P.; Gosai, H.B. Exploring Current Scenario and Developments in the Field of Microbial L-Asparaginase Production and Applications: A Review. Process Biochem. 2022, 121, 529–541.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
23 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No