Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sarfaraz K. Niazi | -- | 4769 | 2023-11-22 04:26:18 | | | |
| 2 | Sirius Huang | Meta information modification | 4769 | 2023-11-23 01:49:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Niazi, S.K.; Magoola, M. mRNA and Synthesis-Based Therapeutic Proteins. Encyclopedia. Available online: https://encyclopedia.pub/entry/51884 (accessed on 09 August 2026).
Niazi SK, Magoola M. mRNA and Synthesis-Based Therapeutic Proteins. Encyclopedia. Available at: https://encyclopedia.pub/entry/51884. Accessed August 09, 2026.
Niazi, Sarfaraz K., Matthias Magoola. "mRNA and Synthesis-Based Therapeutic Proteins" Encyclopedia, https://encyclopedia.pub/entry/51884 (accessed August 09, 2026).
Niazi, S.K., & Magoola, M. (2023, November 22). mRNA and Synthesis-Based Therapeutic Proteins. In Encyclopedia. https://encyclopedia.pub/entry/51884
Niazi, Sarfaraz K. and Matthias Magoola. "mRNA and Synthesis-Based Therapeutic Proteins." Encyclopedia. Web. 22 November, 2023.
Copy Citation
Recombinant technology has been around for nearly three-quarters of a century and has revolutionized protein therapy. However, the cost of developing recombinant therapeutic proteins and the manufacturing infrastructure keep their cost unaffordable for most patients. Proteins are produced in the body via messenger RNA (mRNA) translation. This process can be readily replicated by administering a chemical nucleic acid product to manufacture the same protein recombinantly. The progress made in creating these proteins ex vivo in a cell-free system also offers a lower-cost option to produce therapeutic proteins.
mRNA
therapeutic proteins
recombinant technology
ribosomal translation
cell-free protein expression
1. Introduction
Human cells translate thousands of proteins to maintain bodily functions, including hormones, enzymes, antibodies, and cytokines. The synthesis of these proteins starts with DNA transcription, which leads to the formation of RNA, which is then capped, making mRNA sent out into the cytoplasm to translate into proteins by passing it through the ribosomes. Transcription errors can result in protein deficiency, leading to severe illness. However, little has been achieved except for extracting proteins such as insulin and erythropoietin from animal organs and thousands of liters of human urine. This dilemma was resolved in 1972 when researchers from UC, San Francisco, and Stanford [1] created recombinant cell lines to express proteins; the first such protein was insulin, which was expressed in recombinant E. coli. Currently, over 250 recombinant produced proteins have been approved by the FDA [2]. Developing these proteins as new drugs costs billions of dollars because proteins expressed in recombinant cells do not always match endogenous proteins, require multiple novel post-translational modifications, and their structural variability from batch to batch requires extensive safety and efficacy testing. Their copies as biosimilars cost hundreds of millions for the same reason.
Three quarters of a century after the first recombinant protein was introduced, we experienced a significant event: the first validation of mRNA-based translation of antigenic proteins as COVID-19 vaccines. Although mRNA technology has been known for decades, its delivery and proof of efficacy have only recently been well established, opening doors to many novel applications based on translation rather than the expression of proteins.
2. mRNA Protein Translation
Unlike DNA vaccines, mRNA only works in a certain translational location. This lowers the risks of it getting into the nucleus and causing gene interactions. There are proteins that can stay inside the cell, like neprilysin, rain-derived neurotrophic factor (BDNF), hepatocyte growth factor (HGF), ascular endothelial growth factor (VEGF), and ornithine transcarbamylase (OTC), or they can be on the surface, like cystic fibrosis transmembrane conductance regulator (CFTR) protein. Often, they are sent outside the cell as antigens that represent infectious organisms that will lead to infection.
mRNA, which codes for specific proteins, and non-coding RNAs, including ncRNA, miRNA, and lncRNA, are the two forms of RNA found in cells. The non-coding RNAs, which comprise 98% of all RNAs, ensure that the coding mRNA operates correctly. Since mRNA contains information on the amino acid sequence of the target protein, it serves as a template to produce proteins. Non-coding or untranslated regions of each mRNA molecule regulate how the mRNA is processed.
RNA is converted to mRNA in the nucleus. These modifications, including the 5′ cap and the poly(A) tail, were discovered in the 1950s [3]; many more modifications have been recognized since then to ensure the optimal delivery of the target protein. For example, it was recently discovered that when mRNA is used to produce an antigenic protein, changing a few specific amino acids helps the translated protein collapse after translation and changes its antigenicity through folding incorrectly; this modification was applied to the COVID-19 vaccine [4]. However, these modifications are irrelevant when the translated protein is innate and not antigenic.
Figure 1 shows mRNA structure, and Table 1 lists mRNA designs that can translate any protein through substituting the coding region of the desired protein.

Figure 1. A diagrammatic structure of a typical human protein-coding mRNA, including the untranslated regions (UTRs). It is drawn approximately to scale. The cap is only one modified base. The mRNA cap is a highly methylated modification of the 5′ end of RNA pol II-transcribed RNA. It protects RNA from degradation; recruits complexes involved in RNA processing, export, and translation initiation; and marks cellular mRNA as “self” to avoid recognition by the innate immune system. In eukaryotes, the 5′ cap (cap-0), found on the 5′ end of an mRNA molecule, consists of a guanine nucleotide (G) connected to mRNA via an unusual 5′-to-5′ triphosphate linkage. This guanosine is methylated on the 7 position directly after capping in vivo by a methyltransferase. The average 5′ UTR length is 170, and 3′ UTR length is 700. Polyadenylation is the addition of a poly(A) tail to an RNA transcript, typically an mRNA. The poly(A) tail consists of multiple adenosine monophosphates; it is part of the process that produces mature mRNA for translation; it promotes RNA degradation. Reproduced from http://commons.wikimedia.org/wiki/Image:MRNA_structure.png (accessed on on 1 September 2023).
Table 1. A general plan for mRNA design with an example for granulocyte stimulating factor (G-CSF).
| 5′UTR cap |
| GAGAATAAACTAGTATTCTTCTGGTCCCCACAGACTCAGAGAGAACCCGCCACATGTTCGTGTTCCTGGTGCTGCTGCCTCTGGTGTCCA |
| Start codon (Kozak) |
| GCAGCCAGTGCGTGAACCTGACCACCCGGACCCAGCTGCCACCAGCCTACACCAACAGCTTCACCCGGGGCGTCTACTACCCCGACAAGGT |
| Coding Sequence (CDS) |
| UGGCGGGCCCGGCGACCCAGAGCCCGAUGAAACUGAUGGCGCUGCAGCUGCUGCUGUGGCAUAGCGCGCUGUGGACCGUGCAGGAAGCGACCCCGCUGGGCCCGGCGAGCAGCCUGCCGCAGAGCUUUCUGCUGAAAUGCCUGGAACAGGUGCGCAAAAUUCAGGGCGAUGGCGCGGCGCUGCAGGAAAAACUGGUGAGCGAAUGCGCGACCUAUAAACUGUGCCAUCCGGAAGAACUGGUGCUGCUGGGCCAUAGCCUGGGCAUUCCGUGGGCGCCGCUGAGCAGCUGCCCGAGCCAGGCGCUGCAGCUGGCGGGCUGCCUGAGCCAGCUGCAUAGCGGCCUGUUUCUGUAUCAGGGCCUGCUGCAGGCGCUGGAAGGCAUUAGCCCGGAACUGGGCCCGACCCUGGAUACCCUGCAGCUGGAUGUGGCGGAUUUUGCGACCACCAUUUGGCAGCAGAUGGAAGAACUGGGCAUGGCGCCGGCGCUGCAGCCGACCCAGGGCGCGAUGCCGGCGUUUGCGAGCGCGUUUCAGCGCCGCGCGGGCGGCGUGCUGGUGGCGAGCCAUCUGCAGAGCUUUCUGGAAGUGAGCUAUCGCGUGCUGCGCCAUCUGGCGCAGCCG |
| 3′UTR |
| GCCCCTTTCCCGTCCTGGGTACCCCGAGTCTCCCCCGACCTCGGGTCCCAGGTATGCTCCCACCTCCACCTGCCCCACTCACCACCTCTGCTAGTTCCAGACACCTCCCAAGCACGCAGCAATGCAGCTCAAAACGCTTAGCCTAGCCACACCCCCACGGGAAACAGCAGTGATAACCTTTAGCAATAAACGAAAGTTTAACTAAGCTATACTAACCCCAGGGTTGGTCAATTTCGTGCCAGCCACACCCTGGAGCTAGCA |
| poly-A chain |
The translation process involves an mRNA-coding sequence that passes through the ribosome to create a target protein (Figure 2).
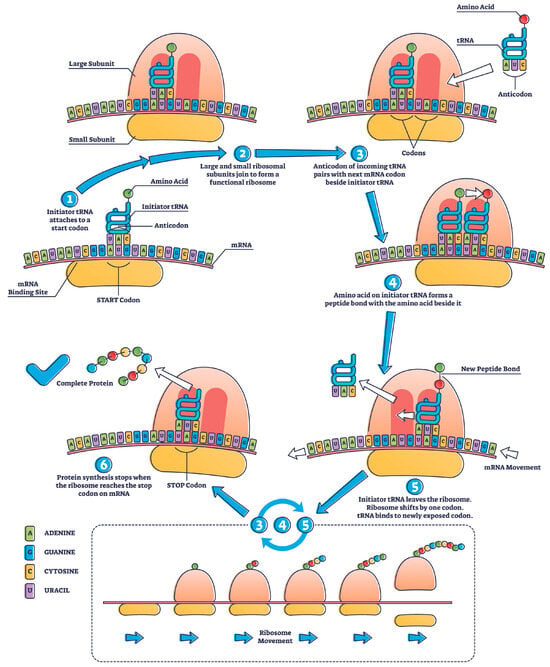
Figure 2. Mechanisms of protein translation in the ribosome [Source: licensed Adobe stock image].
The number of protein molecules translated from one mRNA molecule varies based on the length of the mRNA, translation efficiency, and the stability of the resulting protein. Notably, translation is a dynamic process, and multiple ribosomes can simultaneously translate the same mRNA molecule, forming a chain of ribosomes known as polysomes. A single mRNA molecule can be translated by multiple ribosomes in a process called polysome or ribosome “clustering”. This allows for the efficient and simultaneous production of multiple protein molecules from the same mRNA template. The number of ribosomes that can translate mRNA simultaneously is determined by factors such as ribosome availability, cellular conditions, and specific mRNAs and their associated regulatory elements.
The “translation efficiency” concept significantly determines how many protein molecules can be produced from a single mRNA. Translation efficiency depends on factors such as the presence of translation initiation sites, stability of the mRNA molecule, and availability of various translation factors.
The number of protein molecules translated from one mRNA molecule is not fixed and can vary significantly depending on cellular and molecular factors. Polysomes play a critical role in maximizing protein production, and multiple ribosomes can work together to translate a single mRNA molecule, leading to the generation of multiple protein molecules. For example, polysome or ribosome clustering during translation produces hemoglobin in red blood cells. Hb is a complex protein comprising four subunits: two alpha-globin chains and two beta-globin chains.
While translating globin mRNA into red blood cell precursors (reticulocytes), multiple ribosomes can simultaneously bind to a single mRNA molecule and move along the mRNA simultaneously. This allows for the efficient and rapid production of globin protein subunits to be assembled into functional hemoglobin molecules. Clustering ribosomes on a single mRNA is an example of polysome formation, enhancing the hemoglobin synthesis’s overall translation efficiency [5].
The example in Table 1 for G-CSF can be used to calculate mRNA dose based on the molarity ratio of the translated protein. The dosing is reduced significantly if the mRNA is self-amplifying (saRNA), which is based on the backbone sequence of alphavirus that is a positive-sense single-stranded RNA replicating virus, to allow for prolonged replication; generally, the saRNA required is 10-fold less than the standard mRNA [6]. However, this comparison should consider that standard mRNA can have a poly(A) tail [7] of variable length that is not attached to saRNA, which also prolongs the life of mRNA in the cytoplasm. The size of saRNAs is also larger (9–12 kb) [7].
The regulatory agencies' list of approved therapeutic proteins can be used to find possible targets for using mRNA technology to translate therapeutic proteins instead of expressing them outside of living cells. Table 2 lists the FDA licensed classes of proteins that are eligible as mRNA delivery products. Table 3 lists prominent endogenous proteins that are now manufactured ex vivo, making them an obvious choice.
Table 2. FDA-approved therapeutic protein types [8].
| Therapeutic Protein | Numbers |
|---|---|
| Monoclonal antibody | 94 |
| Hormone | 10 |
| Enzyme | 8 |
| Cytokine | 4 |
| Bispecific antibody | 3 |
| Coagulation factor | 3 |
| Growth factor | 3 |
| Peptide | 3 |
| Carrier protein | 1 |
| Enzyme inhibitor | 1 |
| Fab | 1 |
| Fusion proteins | 1 |
| Single-domain antibody | 1 |
| Toxin | 1 |
Table 3. Endogenous proteins are available as ex vivo-expressed commercial products that can be choice mRNA products.
| Protein | Brand Name (Product) |
|---|---|
| Erythropoietin (EPO) | Epogen, Procrit (Epoetin alfa) |
| Insulin | Humalog, NovoLog (Insulin lispro); Lantus, Levemir (Insulin glargine) |
| Factor VIII | Advate, Kogenate FS, Eloctate (Recombinant factor VIII) |
| Granulocyte-Colony Stimulating Factor (G-CSF) | Neupogen, Neulasta (Filgrastim, Pegfilgrastim) |
| Tissue Plasminogen Activator (tPA) | Activase, Cathflo Activase (Alteplase) |
| Growth Hormones | Genotropin, Humatrope, Norditropin (Somatropin) |
| Interleukins | Proleukin (Aldesleukin) |
| Monoclonal Antibodies | Herceptin (Trastuzumab); Rituxan (Rituximab); Humira (Adalimumab) |
| Enzyme Replacement Therapies | Cerezyme (Imiglucerase); Fabrazyme (Agalsidase beta) |
| Hormone Replacement Therapies | Premarin (Conjugated estrogens); AndroGel (Testosterone) |
Non-endogenous proteins are expressed outside of living cells and can be bought as commercial products (Table 4). They can also be great choices for delivering mRNA. However, the intellectual property infringement issue remains unresolved since there has never been an alternate choice like this before.
Table 4. Non-endogenous recombinant protein candidates for mRNA therapy.
| Therapeutic Protein | Use |
|---|---|
| Abatacept (Orencia) | Rheumatoid arthritis and juvenile idiopathic arthritis |
| Abciximab (ReoPro) | Prevention of platelet aggregation in angioplasty |
| Acalabrutinib (Calquence) | Mantle cell lymphoma and chronic lymphocytic leukemia |
| Adalimumab (Humira) | Various autoimmune disorders |
| Aducanumab (Aduhelm) | Alzheimer’s disease (monoclonal antibody) |
| Aflibercept (Eylea) | Wet age-related macular degeneration |
| Alemtuzumab (Campath) | Chronic lymphocytic leukemia |
| Alpelisib (Piqray) | Breast cancer with PIK3CA mutations |
| Alteplase (Activase) | Thrombolytic drug for acute myocardial infarction, acute ischemic stroke, and PE |
| Anakinra (Kineret) | Rheumatoid arthritis |
| Atezolizumab (Tecentriq) | Various types of cancer |
| Avelumab (Bavencio) | Various types of cancer |
| Basiliximab (Simulect) | Prevention of organ rejection in transplantation |
| Belatacept (Nulojix) | Immunosuppressive therapy for kidney transplantation |
| Belimumab (Benlysta) | Systemic lupus erythematosus |
| Bevacizumab (Avastin) | Various cancers inhibit angiogenesis |
| Bivalirudin (Angiomax) | Anticoagulant for patients undergoing percutaneous coronary intervention |
| Blinatumomab (Blincyto) | Acute lymphoblastic leukemia |
| Cabotegravir (Vocabria) | Long-acting HIV-1 integrase inhibitors for prevention |
| Cabozantinib (Cometriq) | Advanced renal cell carcinoma and hepatocellular carcinoma |
| Certolizumab pegol (Cimzia) | Crohn’s disease, rheumatoid arthritis |
| Cetuximab (Erbitux) | Metastatic colorectal cancer, head and neck cancer |
| Daclizumab (Zinbryta) | Multiple sclerosis |
| Daptomycin (Cubicin) | Antibiotic for complicated skin and skin structure infections |
| Daratumumab (Darzalex) | Multiple myeloma |
| Darolutamide (Nubeqa) | Prostate cancer |
| Denosumab (Prolia, Xgeva) | Osteoporosis and prevention of skeletal-related events in cancer patients |
| Dulaglutide (Trulicity) | Type 2 diabetes |
| Durvalumab (Imfinzi) | Various types of cancer |
| Eculizumab (Soliris) | Paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome |
| Efalizumab (Raptiva) | Psoriasis |
| Elotuzumab (Empliciti) | Multiple myeloma |
| Emapalumab (Gamifant) | Hemophagocytic lymphohistiocytosis (HLH) |
| Eptifibatide (Integrilin) | Antiplatelet drug for acute coronary syndrome |
| Erdafitinib (Balversa) | Urothelial cancer with FGFR mutations |
| Eteplirsen (Exondys 51) | Duchenne muscular dystrophy |
| Fingolimod (Gilenya) | Multiple sclerosis |
| Golimumab (Simponi) | Rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis |
| Golodirsen (Vyondys 53) | Duchenne muscular dystrophy |
| Guselkumab (Tremfya) | Moderate-to-severe plaque psoriasis |
| Gusperimus (Zavesca) | Gaucher disease and Niemann-Pick disease type C |
| Ibalizumab (Trogarzo) | Multidrug-resistant HIV-1 |
| Icatibant (Firazyr) | Hereditary angioedema attacks |
| Inclisiran (Leqvio) | Hypercholesterolemia |
| Infliximab (Remicade) | Autoimmune diseases like Crohn’s and rheumatoid arthritis |
| Ipilimumab (Yervoy) | Melanoma |
| Isatuximab (Sarclisa) | Multiple myeloma |
| Lanadelumab (Takhzyro) | Prevention of hereditary angioedema attacks |
| Laronidase (Aldurazyme) | Enzyme replacement therapy for Hurler syndrome |
| Lenalidomide (Revlimid) | Multiple myeloma and myelodysplastic syndromes |
| Liraglutide (Victoza) | Type 2 diabetes |
| Margetuximab (Margenza) | HER2-positive breast cancer |
| Natalizumab (Tysabri) | Multiple sclerosis and Crohn’s disease |
| Naxitamab (Danyelza) | Neuroblastoma in children |
| Nivolumab (Opdivo) | Various types of cancer |
| Nusinersen (Spinraza) | Spinal muscular atrophy |
| Obiltoxaximab (Anthim) | Inhalational anthrax |
| Obinutuzumab (Gazyva) | Chronic lymphocytic leukemia, follicular lymphoma |
| Ofatumumab (Arzerra) | Chronic lymphocytic leukemia and multiple sclerosis |
| Olaratumab (Lartruvo) | Soft-tissue sarcoma |
| Omalizumab (Xolair) | Asthma and chronic idiopathic urticaria |
| Palifermin (Kepivance) | Prevention of severe oral mucositis in cancer patients |
| Palivizumab (Synagis) | Prevention of respiratory syncytial virus in premature infants |
| Panitumumab (Vectibix) | Metastatic colorectal cancer |
| Panobinostat (Farydak) | Multiple myeloma |
| Pegaspargase (Oncaspar) | Acute lymphoblastic leukemia |
| Pegloticase (Krystexxa) | Refractory gout |
| Pembrolizumab (Keytruda) | Various types of cancer |
| Pemetrexed (Alimta) | Chemotherapy for non-small-cell lung cancer and mesothelioma |
| Pertuzumab (Perjeta) | HER2-positive breast cancer |
| Pexidartinib (Turalio) | Tenosynovial giant cell tumor |
| Plasminogen (Ryplazim) | Congenital plasminogen deficiency |
| Ramucirumab (Cyramza) | Stomach cancer, colorectal cancer, and lung cancer |
| Ranibizumab (Lucentis) | Wet age-related macular degeneration |
| Rituximab (Rituxan) | Non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and vasculitis |
| Rucaparib (Rubraca) | Ovarian cancer with BRCA mutations |
| Secukinumab (Cosentyx) | Psoriasis and ankylosing spondylitis |
| Selinexor (Xpovio) | Multiple myeloma and diffuse large B-cell lymphoma |
| Siltuximab (Sylvant) | Multicentric castleman’s disease |
| Tafasitamab (Monjuvi) | Diffuse large B-cell lymphoma |
| Tildrakizumab (Ilumya) | Moderate-to-severe plaque psoriasis |
| Tislelizumab (Bavencio) | Various types of cancer |
| Tocilizumab (Actemra) | Cytokine release syndrome and rheumatoid arthritis |
| Trastuzumab (Herceptin) | HER2-positive breast cancer |
| Ustekinumab (Stelara) | Psoriasis, psoriatic arthritis, and Crohn’s disease |
| Vedolizumab (Entyvio) | Inflammatory bowel diseases (Crohn’s and UC) |
| Vemurafenib (Zelboraf) | BRAF-mutated melanoma |
| Venetoclax (Venclexta) | Chronic lymphocytic leukemia |
mRNA can also be used for newer biological drugs, regardless of their type or application, as long as their structural sequence is known. However, it does not apply to modified proteins such as pegylated forms or conjugates.
3. Post-Translational Modification (PTM)
The PTMs of proteins are crucial regulatory mechanisms in cellular processes, altering protein structure, function, and localization after translation. Phosphorylation, mediated by kinases, adds phosphate groups to serine, threonine, or tyrosine residues [9]. Glycosylation attaches carbohydrate molecules to asparagine (N-linked) or serine/threonine (O-linked) residues, impacting stability and cell interactions [10]. Acetylation, through acetyltransferases, adds acetyl groups to amino-termini or lysine residues [11]. Methylation, catalyzed by methyltransferases, affects lysine or arginine residues [12]. Ubiquitination, mediated by E3 ligases, marks proteins for degradation or alters localization [13]. SUMOylation adds SUMO proteins to lysines [14]. Prenylation attaches lipid groups to cysteines [15]. Proteolytic cleavage activates precursor proteins, e.g., insulin [16]. Sulfation adds sulfate to tyrosines [17], and ADP-ribosylation can modify proteins in response to cellular stress [18]. Dysregulation of these PTMs is linked to various diseases [19], highlighting their importance in understanding cellular biology and developing targeted therapies.
When proteins are expressed outside of living cells, they go through different post-translational modifications (PTMs) and work less efficiently than proteins that are expressed in living cells, like those made from mRNA. This is because the enzymes and substrates are different, and there are not any specific cell compartments. The availability and variety of enzymes essential for PTMs might be limited in ex vivo settings, leading to incomplete or absent modifications [20]. Similarly, substrate specificity and availability are pivotal; for instance, variations in available sugar donors for glycosylation can yield different glycan structures [21]. The absence of cellular compartments, such as the Golgi apparatus and endoplasmic reticulum in ex vivo systems, particularly cell-free systems, can result in proteins devoid of certain PTMs [22]. Moreover, protein folding, facilitated by chaperone proteins in vivo, might be compromised ex vivo, affecting both the expressed proteins’ structural integrity and associated PTMs [23]. So, while ex vivo expression is important for making large amounts of proteins in a controlled way, getting past PTM problems is the most important thing to do to get proteins that work the same way as proteins expressed in living organisms.
4. mRNA Production
While plasmids continue to be the primary platform for mRNA production, recent research has enabled the production of synthetic mRNA, opening a wide range of prospects for medicinal, gene therapy, and vaccine applications [24]. An oligonucleotide, cDNA made from RNA, a plasmid construct, or the product of a polymerase chain reaction (PCR) are the most common and traditional ways to get a DNA template for in vitro transcription (IVT).
The linearization of DNA is achieved by cleaving the circular DNA at specified locations using restriction enzyme digestion. Additionally, PCR amplification is performed using primers containing the restriction sites of CRISPR-Cas9 and chemical cleavage.
Subsequently, the linearized DNA fragments were purified and analyzed using either DNA sequencing or gel electrophoresis techniques. The incorporation of the 5′UTR, 3′UTR, and poly(A) tail was achieved through the utilization of capping enzymes and poly(A) polymerase, either during the in vitro transcription (IVT) process or after transcription [25]. Lipid nanoparticles (LNP) are used to form a capsule around mRNA molecules to stop them from breaking down, improve cellular absorption, and make sure they get into cells efficiently. The final product was subjected to filtration or gamma-irradiation sterilization, followed by quality control testing. The United States Pharmacopoeia (USP) has made a noteworthy contribution to validating RNA-based products.
Nevertheless, this pertains specifically to mRNA vaccinations. Most of the proposed methodologies apply to various categories of RNA molecules. Moreover, the utilization of the USP approach might lead to a reduction in the expenses associated with method validation, as the USP tests solely require verification [26].
5. mRNA Delivery
Its delivery mode and formulation influence the vaccine efficacy; it is essential for mRNA vaccines because mRNA intracellular delivery presents a significant barrier due to its larger size of 300–1500 kDa, compared to 4–14 kDa for small interference RNA (siRNA) and antisense oligonucleotide (ASO). Another barrier is the cell membrane, which consists of a zwitterionic lipid bilayer and negatively charged phospholipids and repels negatively charged mRNA molecules. mRNA is also susceptible to ribonuclease degradation in the extracellular environment, necessitating robust protection against degradation. Three intracellular barriers exist: endosomal escape, RNA sensors, and endonucleases. When the mRNA-based vaccine reaches the plasma membrane, it is ingested and processed along an endocytic pathway before being released into the cell.
Nevertheless, a small proportion of LNPs evade the endocytic pathway due to the endosomal membrane disruption caused by the protonation of LNP residual amines [27]. This results in the premature release of LNP-mRNA cargo into the cell, thereby diminishing the efficacy of the mRNA vaccine. The recognition of mRNA by cytosolic innate sensors, including Toll-like receptors (TLRs), TLR 3, and TLR7, is an additional barrier to the development of mRNA vaccines. Additionally, intracellular RNases act as a barrier through degrading mRNA before it is translated to produce the antigen in the cell [28].
Numerous methods for delivering mRNA have been developed, including the direct injection of naked mRNA, lipid-based carriers, polymers, and protein derivatives. Compared to other delivery vehicles, lipid nanoparticles have been studied extensively for delivering small molecules, including siRNA and mRNA [29]. Besides their use in SARS-CoV-2 vaccines, LNP-mRNA formulations have also been used to treat genetic disorders, viral infections, and cancer [30].
Vaccines may be administered systemically or locally; the anatomical and physiological properties of the vaccination site, such as the skin, lymphoid organ, or muscle, affect the safety and efficacy of the vaccine. Targeted delivery is also a subject of considerable interest, where the vaccine is injected directly into the target tissue or organ, such as the intranodal injection (Figure 3).
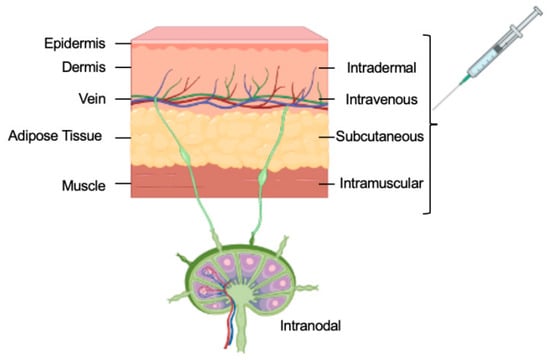
Figure 3. The routes of delivery for mRNA vaccines.
Other delivery methods include intranasal injection and inhalation-based delivery of mRNA vaccines, which are being investigated for respiratory delivery [31]. Inhaled substances encounter pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which induce antigen-presenting cells (APCs) to acquire the antigen. Dendritic cells migrate via lymphatic vessels to lymph nodes, where antigen is presented to naive B and T cells via MHC II complexes. The respiratory system also contains inducible bronchus-associated lymphoid tissue (iBALT), composed of B-cell follicles, plasma cells, T cells, and APCs. Antigens are then presented to both effector B cells and naive T cells [32].
Delivering nucleic acids into cells is the second important area of research [33]. Generally, many routes are available (Figure 3), but an early strategy involved using liposomes composed of phospholipids and cholesterol that resemble cell membranes. Already in 1978, scientists had successfully delivered purified globin mRNA into mouse lymphocytes and human epithelial cells using liposomes [34][35] via entrapping the mRNA within the liposome vesicles. The first cationic lipid (DOTMA) was demonstrated to form stable liposomes when combined with nucleic acids [36]. Positively charged lipids made it easier for negatively charged nucleic acids to stick to them and for them to fuse with negatively charged cell membranes. This made delivery into cells better. The development of cationic lipid-based liposomes (lipofectin) facilitated the delivery of engineered DNA and RNA into cells. Soon after, Lipofectin was used to deliver in vitro-transcribed mRNA into cultured cells to demonstrate protein synthesis [37], paving the way for future therapeutic applications. However, in vivo, applications of lipofectin have shown undesirable side effects, and researchers have continued to seek more effective delivery systems.
Cationic nanoemulsion (CNE) uses cationic lipid nanoemulsion for RNA delivery. The development of ionizable cationic lipids represents a second significant advance. Depending on the pH of the surrounding environment, these lipids may exist in a positively charged or neutral state. It was easier to trap negatively charged mRNA inside the vesicles when these lipid nanoparticles (LNPs) were made at a low pH. However, when delivered in vivo and exposed to physiological pH, the lipids lost their charge, which resulted in several advantages, including a decrease in in vivo toxicity. The delivery of nucleic acids was further optimized through the T-connector, which could generate dense lipid nanoparticles consisting of four components: (i) an ionizable cationic lipid, (ii) a helper lipid, (iii) cholesterol, and (iv) polyethylene glycol (PEG) [38]. Nanoemulsions have both hydrophobic and hydrophilic surfactants that keep the oil core stable in the water phase, which makes particles. Methods such as vigorous agitation, ultrasound, and microfluidics produce nanoemulsions [39]. Figure 4 shows the design of an LNP mRNA product.
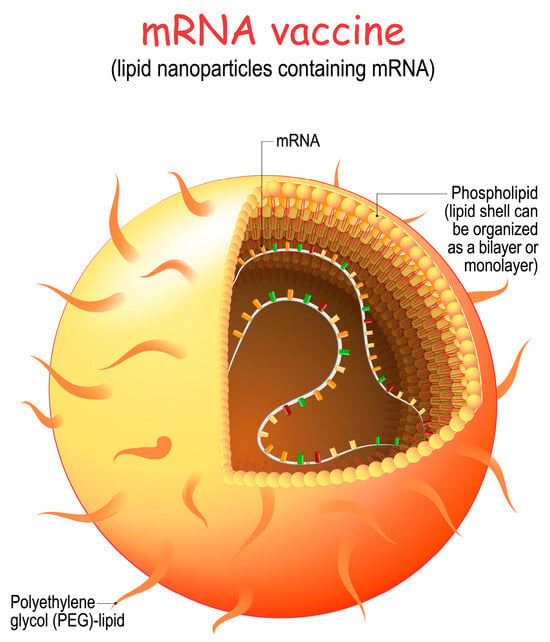
Figure 4. LNP structure of mRNA vaccine (licensed from Shutterstock).
LNP formulation involves the following steps:
-
Lipid Selection: Lipids are carefully selected to form the core structure of the nanoparticle. These lipids are chosen for their ability to self-assemble into nanoparticles and to protect the mRNA from degradation.
-
Encapsulation of mRNA: The synthetic mRNA encoding the target viral protein is mixed with the selected lipids. This mixture is then subjected to microfluidization, or homogenization, which helps encapsulate the mRNA within the lipid nanoparticles.
-
Surface Changes: Polyethylene glycol (PEG) or other molecules can be added to the surface of the lipid nanoparticles to make them more stable, make it harder for the immune system to get rid of them, and make it easier for cells to take them in.
-
Stability and Sterilization: The LNP formulation is rigorously tested to ensure the mRNA remains intact during storage and transportation. The formulation is also sterilized to ensure that it is free from contaminants.
-
Cellular Uptake: After vaccination, the LNPs are injected into the body, where they encounter host cells at the injection site. These LNPs are taken up by antigen-presenting cells such as dendritic cells and macrophages.
-
Translation of mRNA: Once inside the host cells, the mRNA is released from the LNPs and enters the cytoplasm. The cell’s ribosomes then use the mRNA as a template to synthesize the target viral protein, such as the spike protein of the SARS-CoV-2 virus.
-
Immune Response: The newly synthesized viral protein is displayed on the cell’s surface, triggering an immune response. This includes the production of antibodies and activating T cells, which recognize and remember the viral protein.
-
Immune Memory: The immune system “learns” to recognize the viral protein, allowing it to mount a rapid and effective immune response if the actual virus is encountered in the future.
Naked mRNA is administered through dissolving the mRNA in a buffer and injecting the mRNA solution directly into the body. Although naked mRNA cannot diffuse across the intracellular membrane, its internalization mechanism remains unknown. Hydrostatic pressure is hypothesized to lead to the disruption of the cell membrane and facilitate the delivery of nucleic acids into the cytosol [40]. Among the advantages of vaccines based on naked mRNA are storage stability and intrinsic immunogenicity. Upon freeze-drying, naked mRNA can be easily stored at 4 °C for up to 10 months in 10% trehalose [41]. Naked mRNA vaccines are susceptible to RNase degradation and intracellular delivery [42].
Antigen-presenting cells (APCs) are responsible for antigen internalization, processing, and presentation to lymphocytes. Dendritic cells (DCs) are APCs that present processed antigens from microorganisms, tumor cells, and virus-infected cells to T cells to generate an immune response [43]. Due to their migration to T cells in lymph nodes and the high expression of MHC, co-stimulators, and cytokines [44], DCs are suitable vaccination targets. Electroporation disrupts the cell membrane for intracellular nucleic acid delivery through generating an electric shock. Adjusting voltage, capacitance, resistance, and other factors, such as cell number, density, RNA quantity, and pulse time, can improve delivery efficiency. In clinical trials, electroporation has been utilized for DC-based mRNA vaccines [45].
Peptides have been used as vaccine-delivery vehicles. Positively charged amino acid chains, such as lysine and arginine, are necessary for the peptides to function as delivery vehicles. This permits the formation of electrostatic interactions between positively charged peptides and negatively charged mRNA, thereby facilitating the spontaneous formation of complexes [46]. Protamines are advantageous as vaccine carriers for mRNA because they protect it from RNase degradation [47]. The complex of protamine and mRNA has a high adjuvant activity. Due to its structural similarity to the viral RNA genome, the complex is immunogenic upon activation [48]. Like protamines, polymers also prevent mRNA degradation by RNase.
The virus-like packaging and delivery of antigen-encoding, self-amplifying mRNA in the cytoplasm is possible using viral particles. The replicated self-amplifying mRNA can then efficiently express the designated antigens. VRPs are effective in the cytoplasmic delivery of RNA cargo by viral vectors. Viruses internalize and release their genomes into cells via multiple efficient pathways [49]. Single-stranded RNAs, including alphavirus, flavivirus, rhabdovirus, and measles virus, are the most commonly used VRPs for vaccines [50].
The delivery of mRNA to specific cells holds promise for regulating gene expression and potentially addressing genetic mutations or molecular abnormalities associated with various disorders.
5.1. Cell-Based Delivery
In cell-based mRNA delivery, these molecules are therapeutically delivered to the target cells using living cells as carriers or vehicles. This method uses the cells’ built-in capacity to absorb and digest mRNA, enabling therapeutic mRNA’s effective and precise delivery to cells or tissues.
5.2. Extracellular Vesicles
Various cellular types release small membranous structures known as extracellular vehicles (EVs) into the extracellular milieu. The transportation of macromolecules, such as proteins, lipids, and nucleic acids, to neighboring or remote cells plays a vital role in intercellular communication. Recently, electric vehicles (EVs) have exhibited promising capabilities for delivering mRNA, presenting novel opportunities for gene therapy and various other medical applications.
Extracellular vesicles (EVs) serve as natural carriers for mRNA, addressing the abovementioned challenges. Exosomes are non-invasive and scalable ways to deliver mRNA because they come from cells and can be distinguished from bodily fluids like blood, urine, and saliva. The mRNA inside EVs is also protected from being damaged by nucleases, and it stays stable as it moves through the body because it is surrounded by a lipid bilayer membrane.
The utilization of EVs to deliver mRNA offers numerous advantages, primarily due to their ability to selectively target specific cells and locations. EVs can be generated from diverse cell types, including immune, stem, and cancer cells. After that, genetic modification can cause the production of surface proteins or ligands that allow the virus to selectively bind to and enter certain target cells. It is possible to improve the effectiveness and specificity of mRNA delivery by using targeted delivery mechanisms. These mechanisms can also reduce side effects and improve therapeutic outcomes.
5.3. Biomimetic Delivery
Delivering mRNA molecules to specific cells or tissues, called “biomimetic delivery,” employs strategies inspired by natural processes to develop innovative delivery approaches. Using biomimetic mRNA delivery systems could make mRNA-based treatments safer and more effective by getting around the problems with current delivery methods and copying how cells take in mRNA and move it around inside cells.
Using liposomes or lipid nanoparticles that look like the natural lipid bilayer of cell membranes is one way to get biomimetic mRNA distribution to happen. In lipid-based delivery systems, mRNA molecules are enclosed in lipid bilayers. This keeps cells from oxidizing the molecules and helps them get into cells. Liposomes or lipid nanoparticles can also be engineered to mimic a particular cell’s surface features. Consequently, these entities can selectively engage with the desired cells and gain entry through the widely recognized cellular internalization process called endocytosis. The endosomal membrane could be combined with liposomes or lipid nanoparticles to make it easier for the mRNA payload to be released into the cytoplasm, where it can be translated into proteins.
5.4. Tissue Targeting
For mRNA treatment to achieve optimal efficacy, there is a pressing need to develop more sophisticated in vivo delivery systems. The substantial organs, such as the heart, kidneys, brain, and lungs, play a vital role in the functioning of the human body. Regarding ease of administration, the liver is widely regarded as the preferred organ for most molecular medications.
The presence of fenestrated vasculature enables the effective delivery of uniform medicines and allows for the transport of larger particles.
Therefore, simple intravenous delivery makes it easier for the hepatocytes to produce the mRNA cargo effectively, ultimately leading to therapeutic protein levels. More effective delivery methods are needed when an organ other than the liver is the goal. These include employing catheters to deliver medication to the organ or developing delivery methods that target neurons exploiting HSV tropism for neural cells [51][52].
References
- Cohen, S.N.; Chang, A.C.; Boyer, H.W.; Helling, R.B. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244.
- FDA-Licensed Biological Products. Inxight Drugs. National Center for Advancing Translational Sciences. Available online: https://drugs.ncats.io/ (accessed on 9 August 2023).
- Davis, F.F.; Allen, F.W. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957, 227, 907–915.
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840.
- Wu, C.C.; Keck, J.G. The clustering of ribosomes on homogeneous mRNA in the erythrocyte cell-free system. J. Biol. Chem. 1982, 257, 4259–4264.
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129.
- Li, M.; Wang, Z.; Xie, C.; Xia, X. Advances in mRNA vaccines. Int. Rev. Cell Mol. Biol. 2022, 372, 295–316.
- US Licensed Biological Products. Inxight Drug Database. Available online: https://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1 (accessed on 25 April 2023).
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934.
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009.
- Kouzarides, T. Acetylation: A Regul. Modif. Rival Phosphorylation? EMBO J. 2000, 19, 1176–1179.
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849.
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1695, 55–72.
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956.
- Bergo, M.O.; Leung, G.K.; Ambroziak, P.; Otto, J.C.; Casey, P.J.; Young, S.G. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 2001, 276, 9673–9677.
- Steiner, D.F.; Cunningham, D.; Spigelman, L.; Aten, B. The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill Professional: New York, NY, USA, 2002; pp. 962–1009.
- Turnbull, J.E.; Field, R.A.; Brown, J.R. Aliphatic hydroxylation of tyrosine residues in fibrinogen and fibrin: Evidence for the formation of 3, 4-dihydroxyphenylalanine. Biochem. J. 2001, 356, 13–16.
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219.
- Hunter, T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 2007, 28, 730–738.
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252.
- Imperiali, B.; O’Connor, S.E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 1999, 3, 643–649.
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408.
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332.
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 62813.
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl (3′-deoxy)GpppG. RNA 2001, 7, 1486–1495.
- mRNA Vaccines. USP. Available online: https://www.usp.org/mrna (accessed on 15 August 2023).
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334.
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594.
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094.
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41.
- Heida, R.; Hinrichs, W.L.; Frijlink, H.W. Inhaled vaccine delivery in the combat against respiratory viruses: A 2021 overview of recent developments and implications for COVID-19. Expert Rev. Vaccines 2021, 21, 957–974.
- Silva-Sanchez, A.; Randall, T.D. Role of iBALT in Respiratory Immunity. In Inducible Lymphoid Organs; Kabashima, K., Egawa, G., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 21–43.
- Nitika, J.W.; Hui, A.M. The Delivery of mRNA Vaccines for Therapeutics. Life 2022, 12, 1254.
- Dimitriadis, G.J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 1978, 274, 923–924.
- Ostro, M.J.; Giacomoni, D.; Lavelle, D.; Paxton, W.; Dray, S. Evidence for translation of rabbit globin mRNA after liposome-mediated insertion into a human cell line. Nature 1978, 274, 921–923.
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21.
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081.
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res. 2005, 22, 362–372.
- Ganta, S.; Talekar, M.; Singh, A.; Coleman, T.P.; Amiji, M.M. Nanoemulsions in translational research-opportunities and challenges in targeted cancer therapy. AAPS PharmSciTech 2014, 15, 694–708.
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531.
- Jones, K.L.; Drane, D.; Gowans, E.J. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques 2007, 43, 675–681.
- Singer, D.F.; Linderman, J.J. The relationship between antigen concentration, antigen internalization, and antigenic complexes: Modeling insights into antigen processing and presentation. J. Cell Biol. 1990, 111, 55–68.
- Mbongue, J.; Nicholas, D.; Firek, A.; Langridge, W. The role of dendritic cells in tissue-specific autoimmunity. J. Immunol. Res. 2014, 2014, 857143.
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593.
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909.
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896.
- Stitz, L.; Vogel, A.; Schnee, M.; Voss, D.; Rauch, S.; Mutzke, T.; Ketterer, T.; Kramps, T.; Petsch, B. A thermostable messenger RNA based vaccine against rabies. PLoS Negl. Trop. Dis. 2017, 11, e0006108.
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkic-Zrna, S.; Probst, J.; Kallen, K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15.
- Vázquez-Calvo, Á.; Saiz, J.-C.; McCullough, K.C.; Sobrino, F.; Martín-Acebes, M.A. Acid-dependent viral entry. Virus Res. 2012, 167, 125–137.
- Lundstrom, K. Replicon RNA Viral Vectors as Vaccines. Vaccines 2016, 4, 39.
- Grankvist, R.; Jensen-Urstad, M.; Clarke, J.; Lehtinen, M.; Little, P.; Lundberg, J.; Arnberg, F.; Jonsson, S.; Chien, K.R.; Holmin, S. Superselective endovascular tissue access using trans-vessel wall technique: Feasibility study for treatment applications in heart, pancreas and kidney in swine. J. Intern. Med. 2019, 285, 398–406.
- Mali, S. Delivery systems for gene therapy. Indian J. Hum. Genet. 2013, 19, 3–8.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
23 Nov 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No