1. Introduction
The modular role of tiny protein domains has emerged as a new subject in signal transduction biology and protein biochemistry. These can, in certain situations, directly control catalytic activity. In others, they function to bind essential regulatory proteins. Some of the most well-studied models are SH2 (src homology 2) domains
[1]. In 1994, scientists discovered a phosphorylated protein linked to the protein Shc1 after BCR or cytokine receptor activation
[2]. It has two significant isoforms: SHP1 and SHP2
[3]. SHP2 interacts with its targets via its SH2 domains, and binding via both SH2 domains results in its maximal enzymatic function
[4].
SHP2 is also a well-known modulator of multiple other signaling pathways, making it an appealing anti-cancer therapeutic target
[5]. Various cancer cell signaling pathways that promote cancer development, such as receptor tyrosine kinases (RTKs), MAPK, PI3K-AKT-mTOR, and JAK-STAT, play a role in making immune checkpoint therapy less effective. These pathways involve the participation of SHP2
[6]. Moreover, immune checkpoint therapy resistance also arises from immune-suppressive signaling pathways like PD-1 and CTLA-4 signaling, in which SHP2 is implicated
[7]. SHP2 influences the tumor microenvironment (TME) by promoting immunosuppressive factors, including cytokines and chemokines, which create an unfavorable immune response environment. Additionally, SHP2 affects TGF-beta signaling, contributing to cancer progression through enhanced epithelial-mesenchymal transition (EMT) and immunosuppression in the TME
[8]. SHP2’s impact extends to myeloid cells, where its interaction with the PD-1-SHP2 axis affects GM-CSF-mediated protein phosphorylation, influencing myeloid differentiation and potentially offering opportunities for cancer immunotherapy
[9].
2. The Structure of SHP2
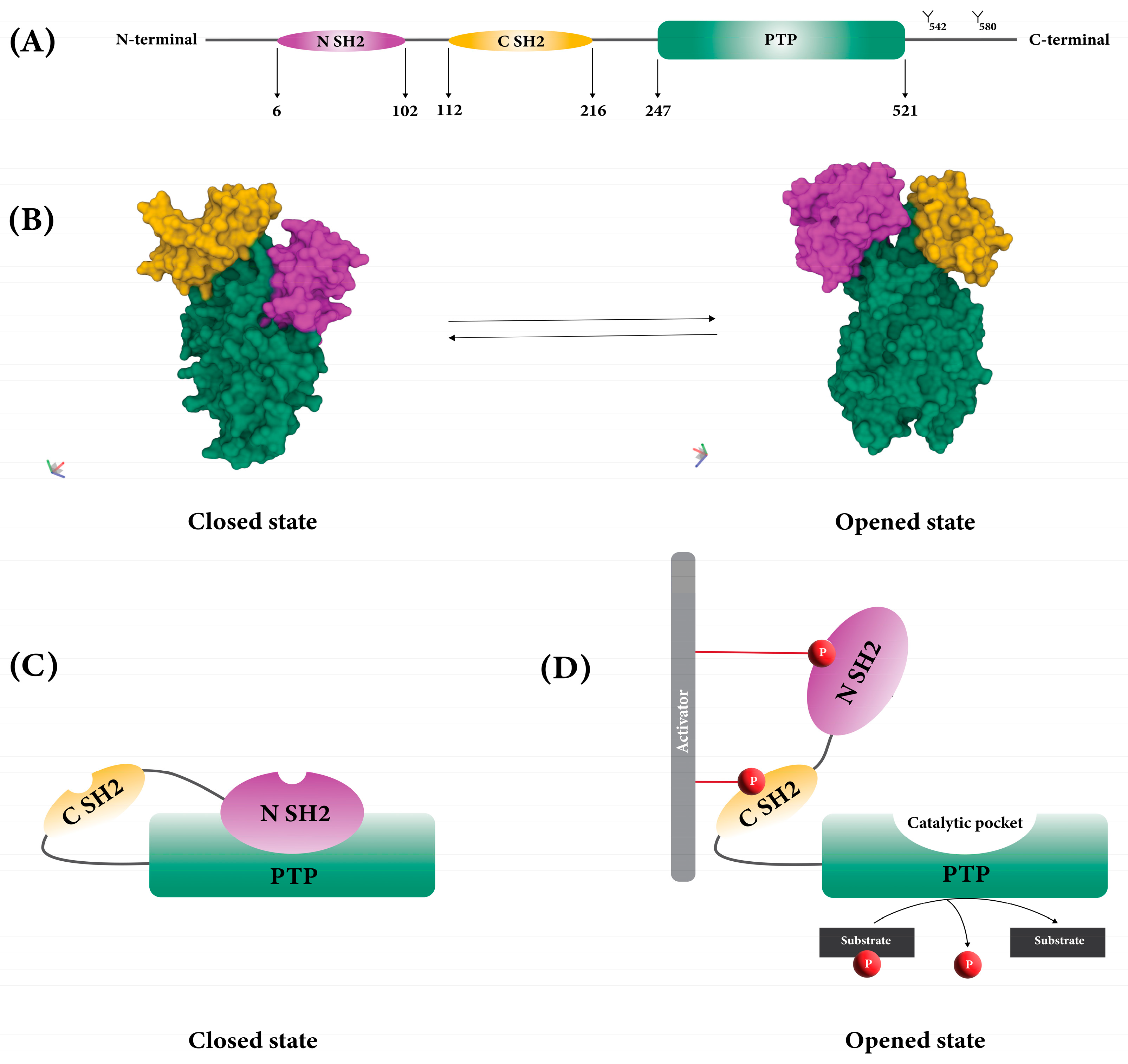
SHP2 consists of two SH2 domains (called N-SH2 and C-SH2), a protein tyrosine phosphatase domain with catalytic activity (PTP domain), a C-terminal tail with two tyrosine phosphorylation sites (Y542 and Y580), and a proline-rich motif (
Figure 1A)
[10].
Figure 1. Structure of SHP2. (A) Two SH2 domains and PTP domain of SHP2. (B) 3D structure of SHP2 in opened (PDB: 6CRF) and closed (PDB: 5EHR) states. (C) Auto-inhibition of SHP2 due to the blocking of the PTP catalytic site by N-SH2. (D) Exposure of the catalytic pocket due to the conformational change of SHP2.
The function of SHP2 is regulated by its structural conformation between closed (or basal) and opened states (
Figure 1B)
[11]. When SHP2 is in its closed position, auto-inhibition happens because the back of the N-SH2 domain forms a loop and interacts intramolecularly with PTP, which has a catalytic pocket. This interaction blocks the catalytic pocket and stops SHP2’s substrates from getting to it (
Figure 1C)
[10][11].
When growth factors and cytokines stimulate cells, SH2 domains bind to phosphorylated tyrosine residues on various proteins, including cytokine receptors, receptor tyrosine kinases, and scaffolding adaptors. Once a phosphotyrosyl peptide binds to the N-SH2 domain, SHP2 undergoes a conformational change, which exposes its catalytic pocket. This open, active conformation allows the SHP2 substrates to access the catalytic pocket and leads to dephosphorylation (
Figure 1D)
[11][12][13].
3. Immune Checkpoints
Immune checkpoints are a multitude of natural inhibitory pathways ingrained within the immune system. These pathways are vital in preserving self-tolerance and regulating the extent and duration of immune responses in peripheral tissues
[14]. Different immune cells, including T cells, NK cells, macrophages, and dendritic cells, express various immune checkpoint receptors on their surface. Each of these immune cells has distinct ligand molecules specifically expressed in tumor cells
[15]. Immune checkpoints, particularly CTLA-4, PD-1, and PD-L1, can negatively regulate T cells by activating SHP2, leading to immune tolerance and allowing tumors to evade immune surveillance
[16][17][18].
4. Immune Checkpoint Therapy
Immune checkpoint therapy is a revolutionary approach to cancer treatment that harnesses the body’s immune system to target and eliminate cancer cells. It involves the use of monoclonal antibodies that block the inhibitory signals generated by immune checkpoints, thereby reinvigorating the antitumor immune response and unleashing the full potential of T cells to recognize and attack cancer cells. By releasing the brakes on the immune response, this therapy enhances the immune system’s ability to detect and destroy tumors
[19]. While well-studied immune checkpoints like PD-1, CTLA-4, and PD-L1 have shown clinical benefits, an expanding range of other immune checkpoints are being investigated for their potential benefits
[20].
5. Immune Checkpoint Therapy Resistance Mechanisms
Immune checkpoint therapy, while often effective, can face challenges due to the development of resistance mechanisms. Several factors contribute to immune checkpoint therapy resistance, including:
5.1. TME Changes
The alterations within TME can lead to immune checkpoint inhibition and resistance through various mechanisms. Immunosuppressive cytokines, regulatory T cells, myeloid-derived suppressor cells (MDSCs), exhausted T cells and molecules like indole-2,3-dioxygenase (IDO) contribute to an immunosuppressive environment. Factors intrinsic to tumor cells, including mutational load, oncogenic signaling pathways, PD-L1 expression, and MHC class 1 downregulation, also play a role. The elevated secretion of inhibitory cytokines such as IL-6, IL-10, and TGF-beta by tumor cells contributes to immune tolerance, hindering cytotoxic T-cell function and promoting Treg and MDSC infiltration. High expression of immune suppressive molecules like IDO and PD-L1 counters immune activation even when CD8-T cells are present. Tryptophan catabolism by IDO leads to immunosuppressive metabolites that inhibit T-cell expansion. PD-L1 expression by tumor cells also influences immune escape
[21][22].
5.2. Loss of Antigen Expression
Cancer cells may downregulate the expression of antigens that T cells recognize. Any process that contributes to the reduction in the presentation and expression of tumor antigens can result in the development of acquired resistance to checkpoint inhibitors. Tumors that undergo immune editing and select low-immunogenicity sub-clones with reduced neo-antigen expression can become resistant to checkpoint inhibitors. Strategies to overcome the lack of tumor immunogenicity include adoptive cell therapy (ACT) with T-cells targeting specific antigens, creating mutational neo-antigens through vaccinations, and using epigenetic drugs to increase tumor antigen expression
[23].
5.3. New Genetic Mutations
Gene mutations in both tumor cells and immune cells within the TME can lead to a situation where cancer cells evade recognition by T cells and can influence the response to immune checkpoint blockade (ICB) therapy. Tumor cells and immune cells interact in the cancer microenvironment through inhibitory signaling molecules, and the release of immune suppression by ICBs activates the immune system, leading to an inflammatory response at tumor sites. Tumor gene mutations, such as those in the RAS/Raf/MEK/MAPK pathway, EGFR, and JAK genes, have been linked to ICB response. Additionally, high tumor mutational load and low intratumoral mutational heterogeneity have been correlated with better responses to ICBs. However, while promising, the field is still developing, and further research is needed to fully understand the mechanistic basis for the relationship between immune checkpoint therapy resistance and new genetic mutations
[20].
5.4. Upregulation of Inhibitory Molecules
Tumor cells might increase the expression of additional inhibitory molecules beyond the immune checkpoints targeted by therapy, further dampening T-cell responses. A resistance mechanism observed in tumor cells involves the elevation of alternative checkpoint inhibitors in response to monoclonal antibody therapy. For instance, in the context of investigating melanoma or prostate cancer, it was discovered that tumors that initially increased their levels of CTLA-4 and were later treated with anti-CTLA-4 antibodies subsequently raised their VISTA expression. This resulted in the activation of a distinct pathway for suppressing T-cell activity. Tumor cells can adapt by upregulating other immune checkpoints, such as VISTA, when one checkpoint is targeted, leading to alternative pathways for suppressing T-cell responses
[22].
5.5. Upregulation of Cancer Promoting and Immunosuppressive Signaling Pathways in Cancer and Immune Cells Which Involve in ICI Resistance
Resistance to immune checkpoint therapy can arise from the activation of alternative cancer-promoting signaling pathways and the upregulation of additional immunosuppressive signaling pathways
[21]. Numerous oncogenic signaling pathways within cancer cells contribute to resistance against immune checkpoint therapy, including receptor tyrosine kinases (RTKs), MAPK, PI3K-AKT-mTOR, JAK-STAT, Hippo, and Wnt pathways
[6]. Suppressive functions of immune checkpoints usually depend on ligand-induced signaling. There are several immune-suppressive signaling pathways involved in immune checkpoint therapy resistance, including PD-1 signaling, CTLA-4 signaling, TIM3 signaling, LAG3 signaling, TIGIT signaling
[7], IDO (Indoleamine 2,3-dioxygenase) pathway
[24], A2AR (Adenosine A2A Receptor) signaling pathway
[10][25], and CD73-adenosine pathway
[11].
6. Role of SHP2 in Checkpoint Therapy Resistance in Immune and Cancer Cells
6.1. The Immune Suppressive Effects of SHP2 in the TME
SHP2 can affect the TME by promoting immunosuppressive factors, such as cytokines and chemokines, that create an environment less conducive to effective immune response. SHP2’s role in TME extends to modulating TGF-beta signaling, a crucial pathway in cancer progression. SHP2 can enhance TGF-beta-induced epithelial-mesenchymal transition (EMT) and contribute to immunosuppression within the TME
[8]. Indeed, IL-10, an anti-inflammatory cytokine, has an active role in the enhanced antitumor immunity induced by SHP2 or PD-1 targeting in myeloid cells. SHP2 and the PD-1-SHP2 signaling interfere with GM-CSF-mediated phosphorylation of specific proteins, affecting myeloid differentiation. The presence of the PD-1-SHP2 axis in myeloid cells is akin to its established role in B and T lymphocytes. This myeloid-specific PD-1-SHP2 axis could be critical in cancer, where cancer cells can stimulate the production of growth factors and increase PD-1 and PD-L1 expression in myeloid progenitors. Moreover, SHP2 ablation resulted in differentiated neutrophils and tumor-associated macrophages (TAMs) with enhanced effector characteristics, thus promoting antitumor immune responses. Ultimately, SHP2 appears to play a significant role in orchestrating myeloid cell responses within the TME, affecting antitumor immunity and potentially offering avenues for cancer immunotherapy strategies
[9].
The immune system fights tumors by producing IFN-γ (a cytokine that stimulates the activation of immune cells) and activating T cells
[26]. The inhibitory receptor PD-1 hinders T-cell activation by enlisting the enzyme SHP2
[9]. It is shown that blocking SHP2 has the potential to trigger T-cell help (Th1) immunity, stimulate T-cell activity and eliminate the immunosuppressive effect of cancer
[27]. In addition, SHP2 is associated with the skewing of tumor-infiltrating T cells, particularly leukemia Tc1/Th1 cells, towards an inhibitory phenotype, suggesting its involvement in immune regulation within the TME
[28].
In natural killer (NK) cells, cell surface inhibitory receptors that activate SHP2 through the ITIM motif were found
[29]. Furthermore, SHP2-deficient NK cells produced more IFN-γ in response to tumor target cells
[30].
In macrophages, SHP2 regulates multiple signaling pathways, including the CSF-1/CSF-1R axis and CD47/SIRPα, promoting tumor growth and immune evasion in TME. Inhibiting SHP2 in macrophages may promote M1 TAM (antitumor phenotype) polarization and create a microenvironment conducive to antitumor immunity, making it a potential target for tumor immunotherapy
[31][32].
SHP2’s potential contribution to shaping the tumor microenvironment (TME) to be less receptive to immune checkpoint therapies. SHP2’s role in fostering immunosuppressive factors could establish an environment that hampers the effectiveness of immune checkpoint inhibitors, contributing to therapy resistance. The promotion of immunosuppressive factors by SHP2 may hinder the desired immune response triggered by immune checkpoint inhibitors, highlighting the intricate interplay between SHP2-mediated TME modulation and immune checkpoint therapy outcomes. Altogether, SHP2 plays a crucial role in maintaining an immunosuppressive microenvironment by suppressing T-cell activation and enhancing the activation of tumor-promoting M2 macrophages.
6.2. The Role of SHP2 in Tumor Antigen Presentation in Cancer Cells
SHP2 can impact antigen presentation by affecting MHC expression on tumor cells, potentially limiting their recognition by immune cells. Inhibiting SHP2 in cancer cells enhanced the signaling of IFNγ, leading to a rise in the expression of its downstream targets, such as chemoattractant cytokines and antigen-presenting machinery. Moreover, the genetic removal of PTPN11 within tumor cells led to an increased presence of MHC class I molecules in co-culture scenarios. Conversely, introducing a drug-resistant SHP2 mutant into tumor cells did not elevate MHC class I levels when treated with SHP099, confirming that the heightened IFNγ signaling induced by SHP099 in cancer cells is directly attributable to the targeted inhibition of SHP2. Since deficiencies in presenting antigens are linked to resistance against T-cell-triggered tumor elimination, the enhancement of MHC class I expression on tumor cells through SHP2 inhibition offers a valid rationale for synergizing the SHP2 blockade with immunotherapy for cancer patients
[33].
6.3. Role of SHP2 in Tumor-Promoting and Immunosuppressive Signaling Pathways in Cancer and Immune Cells
6.3.1. Immune Roles of SHP2 in Immunosuppressive Signaling Pathways
SHP2 in PD-1 Signaling, RAS/ERK and PI3K/AKT Signaling
PD-1 is crucial in dampening immune reactions and fostering self-tolerance by regulating T-cell function, triggering apoptosis of T cells specific to antigens, and restraining the apoptosis of regulatory T cells
[34]. Within the PD-1 signaling pathway, SHP2 plays a pivotal role by participating in intricate regulatory mechanisms that modulate T-cell receptor (TCR) signaling and downstream events. PD-1 engagement orchestrates the recruitment of SHP2 in proximity to the T-cell receptor, leading to the inhibition of proximal kinase activation and subsequent attenuation of Lck-mediated phosphorylation of ZAP-70. This interaction results in the dampening of key signaling cascades, particularly the PI3K-Akt and Ras-MEK-ERK pathways.
The inhibition of SHP2 within the PD-1 signaling pathway has emerged as a promising strategy in the context of immune checkpoint inhibitor (ICI) resistance therapy. SHP2, a critical regulator of PD-1 signaling, plays a dual role in immune response modulation: it can dampen T-cell activation by interfering with TCR and co-stimulatory signaling while also stimulating TCR signaling through LCK activation. This dual role underscores the complexity of SHP2’s involvement in immune regulation. Leveraging SHP2 inhibition as a therapeutic approach involves addressing its context-dependent functions. In the case of ICI resistance, targeting SHP2 might counteract the dampening effect on T-cell activation and enhance the response to checkpoint inhibitors. However, the precise modulation of SHP2’s inhibitory and stimulatory effects will be crucial to avoid potential adverse effects. As research continues to elucidate the intricate mechanisms of SHP2’s involvement in the PD-1 pathway, novel therapeutic strategies that harness its potential could pave the way for more effective ICI resistance therapies, offering renewed hope for cancer patients facing limited treatment options.
SHP2 in CTLA-4 Signaling
SHP2 emerges as a significant mediator in the CTLA-4 signaling pathway, contributing to the intricate regulation of T-cell activation. Upon T-cell activation, the phosphorylated YVKM motif in CTLA-4’s cytoplasmic tail is implicated in recruiting SHP2, which in turn aids in repressing T-cell activation. This recruitment of SHP2 potentially forms a part of CTLA-4’s mechanism for delivering inhibitory signals. The phosphorylated YVKM motif of CTLA-4 recruits SHP2 to inhibit RAS. CTLA-4 also inhibits AKT activity through PP2A. This finding underscores SHP2’s dual role in immune regulation, as observed in other signaling pathways. While the exact extent of SHP2’s involvement in the CTLA-4 pathway is still being elucidated, its potential interaction with CTLA-4 highlights its significance as a common regulatory element across various immune checkpoint pathways. The interplay between CTLA-4, SHP2, and other associated molecules in orchestrating immune responses adds another layer of complexity to the modulation of T-cell activation and the broader context of immunoregulation
[7][35].
Targeting SHP2 inhibition within the CTLA-4 signaling pathway holds potential for overcoming immune checkpoint inhibitor (ICI) resistance. By disrupting the interplay between SHP2 and CTLA-4, this approach aims to enhance T-cell activation and counteract inhibitory signaling, offering a strategy to restore ICI responsiveness. However, careful modulation is crucial to balance immune activation while preventing potential adverse effects, highlighting SHP2’s significance in designing effective therapies for ICI resistance.
SHP2 in BTLA Signaling
Within the BTLA (B- and T-lymphocyte attenuator) signaling pathway, SHP2’s role is intertwined with the intricate mechanisms governing immune regulation. BTLA, possessing ITIM and ITSM motifs in its cytoplasmic domain, employs these motifs to orchestrate inhibitory signaling. Upon ligand engagement, both tyrosine residues within these motifs become phosphorylated, facilitating the recruitment of SHP1 and SHP2. Notably, BTLA exhibits a preference for recruiting the potent phosphatase SHP1, distinguishing it from PD-1 signaling, which mainly recruits SHP2. This unique recruitment of SHP1 by BTLA contributes to the effective dampening of TCR and CD28 signaling, ultimately regulating T-cell responses. Furthermore, BTLA’s role extends to T follicular helper (Tfh) cells, where its engagement with HVEM on B cells leads to the recruitment of SHP1, culminating in the inhibition of TCR signaling and modulation of B cell proliferation. This intricate interplay underscores SHP2’s role in fine-tuning immune responses through the BTLA pathway, shedding light on potential avenues for immune regulation modulation
[7].
By disrupting SHP2-mediated inhibitory signaling downstream of BTLA engagement, this strategy aims to enhance T-cell activation and counteract resistance mechanisms, potentially restoring ICI effectiveness. Careful modulation of SHP2 activity holds the potential for overcoming ICI resistance, offering a novel avenue for improving therapeutic outcomes.
SHP2 in TCR Signaling and RAS/MAPK Signaling Pathways
T-cell receptor (TCR) signaling leads to T-cell activation, proliferation, and the immune response. SHP2, a critical player in TCR signaling, has a complex role in the pathway, exhibiting both positive and negative regulatory effects depending on the context and signaling molecules involved. Acting as a dual-specificity phosphatase, SHP2 modulates the phosphorylation status and activity of key molecules, such as Zap70 and Lck, integral to TCR-mediated signaling. It can positively regulate TCR signaling by activating downstream molecules like Ras-ERK, which is crucial for T-cell proliferation and differentiation. However, SHP2 also negatively regulates the pathway either due to direct dephosphorylating signaling molecules such as ZAP-70, a key kinase in TCR signaling, or due to the inhibition of TCR signaling via PD-1. This multifaceted involvement of SHP2 shapes T-cell activation and immune responses. In addition, SHP2 contributes to immune regulation via inhibitory receptors. It interacts with inhibitory receptors like PD-1 and BTLA through ITIMs and ITSM, adding complexity as its inhibitory role intersects with positive contributions to TCR signaling. SHP2 deficiency in T cells can lead to impaired TCR signaling and defective immune responses. The interplay of SHP2 with PD-1, including feedback mechanisms, and its broader impact on TCR signaling and immune regulation continue to be active areas of research, influencing the understanding of T-cell responses, especially in the context of cancer immunotherapy
[3][36][37].
SHP2 in BCR Signaling
When PD-1 is co-engaged with BCR, it results in the phosphorylation of both tyrosine residues in PD-1. SHP2 is then recruited to the C-terminal phosphotyrosine of PD-1 and is subsequently phosphorylated. The phosphorylated SHP2 then acts to dephosphorylate proximal signal transducers of BCR, including molecules like Syk and Igα/β. This deactivation of downstream molecules, such as PI3K, PLCγ2, and ERK, leads to inhibiting acute-phase reactions like Ca
2+ mobilization and long-term effects, such as growth retardation. This suggests that SHP2 plays a direct role in regulating BCR signaling through its involvement in the PD-1 pathway, contributing to the modulation of B-cell activation and responses
[38].
SHP2 in NK Cell Signaling
It is shown that SHP2 is associated with NK (natural killer) cell signaling, and SHP2 expression negatively regulates NK cell function. One study describes a newly identified role for SHP2 in dampening NK cell activation, especially in response to tumor target cells. Using various experimental approaches involving knockdown and overexpression of SHP2 in NK-like cell lines, the study demonstrates that SHP2 suppresses cytolytic activity and cytokine production in a concentration-dependent manner. This inhibitory effect impacts MTOC (microtubule organizing center) polarization and granzyme B release during NK cell responses to target cells. Interestingly, SHP2 overexpression reduced NK-target cell conjugation, while SHP2 silencing did not influence this process. The findings highlight SHP2’s role as a general inhibitor of NK cell responsiveness, shedding light on its involvement in regulating NK cell functions and potential implications for therapeutic interventions targeting SHP2 to enhance NK cell activity in cancer or viral infections
[39].
TLR Signaling
SHP2 is directly associated with TLR (Toll-like receptor) signaling. Belonging to the SH2-domain-containing family of non-membrane protein tyrosine phosphatases, SHP2 shares sequence identity with SHP-1 but has ubiquitous expression and regulates diverse signaling pathways. Notably, SHP2 has been found to inhibit IFN production in response to TLR3 and TLR4 ligands, as demonstrated by An et al. SHP2 deficiency led to enhanced IFN-β expression upon TLR activation, with direct interaction between SHP2 and the kinase domain of TBK1 inhibiting IRF3 activation and IFN production. Xu et al. also confirmed that SHP2
−/− macrophages displayed increased IFN-β secretion upon TLR activation. Furthermore, SHP2 was shown to modulate MAPK pathways, with its deletion attenuating JNK and p38 MAPK activation in response to TLR stimulation in various cell types. The exact mechanisms underlying these interactions warrant further investigation, possibly involving TRAF6 ubiquitination and TAK1 activity modulation. Additionally, evidence suggests that SHP2 might be implicated early in TLR engagement, as active SHP2 mutant expression in endothelial cells correlated with reduced LPS-induced barrier disruption and increased FAK phosphorylation
[40].
SHP2 in JAK/STAT Pathway in Cytokine Receptor Signaling
The JAK/STAT signaling pathway, which plays a crucial role in immune cells, regulates gene expression and mediates responses to various cytokines and growth factors, thus significantly influencing immune function and inflammation. This pathway also orchestrates the immune system, particularly in the polarization of T helper cells, and is tightly controlled by proteins like Suppressors of Cytokine Signaling (SOCS), Protein Tyrosine Phosphatases (PTPs), and Protein Inhibitors of Activated STATs (PIAS). Dysregulation of this pathway can lead to various immune disorders. Among the regulators, SOCS proteins, SHP1 and SHP2, negatively modulate JAK-STAT activity through mechanisms such as direct dephosphorylation of JAKs or competitive binding to phosphorylated receptors. These regulators ensure precise initiation, duration, and termination of the signaling cascade, preventing uncontrolled immune responses. The balance of these regulators finely tunes the immune response, and understanding their roles opens avenues for potential therapeutic interventions in immune-related diseases. Additionally, other post-translational modifications like SUMOylation can further influence the activity of STAT proteins. Understanding these regulatory mechanisms offers the potential for targeted therapeutic interventions to treat immune-related diseases and disorders
[41].
6.3.2. Non-Immune Roles of SHP2 in Oncogenic Pathways (Tumor-Promoting) in Cancer Cells
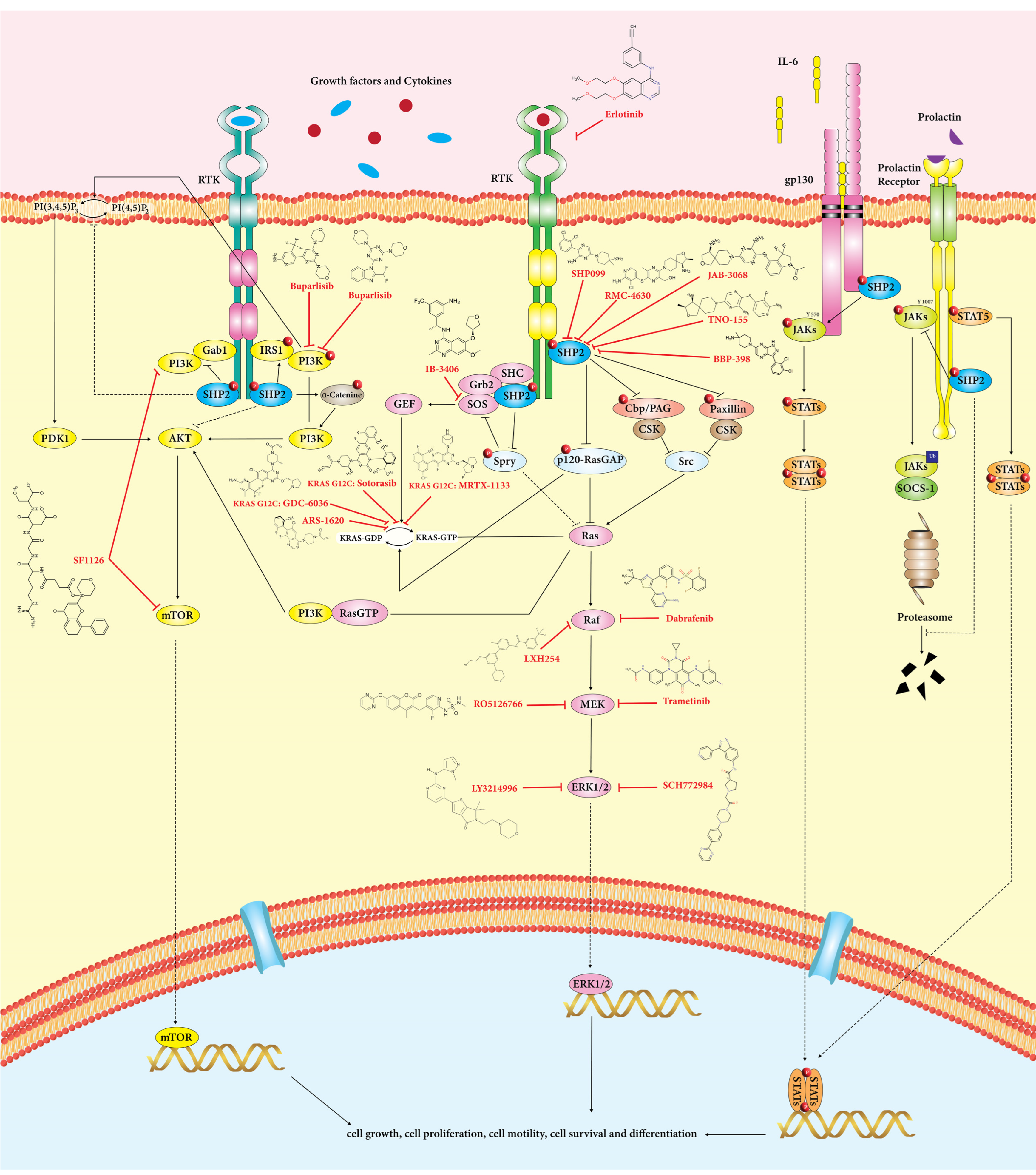
SHP2 participates in several oncogenic signaling cascades, including RAS/MAPK, PI3K/AKT, and JAK/STAT pathways (
Figure 2)
[5][27][42]. In addition, MAPK, PI3K-AKT-mTOR, and JAK-STAT pathways in cancer cells play a crucial role in ICI resistance
[6]
Figure 2. SHP2 is critical in regulating RAS/ERK, PI3K/Akt, and JAK/STAT signaling pathways in cancer cells. These pathways involve cell growth, differentiation, metabolism, and apoptosis. SHP2 positively influences the RAS/ERK signaling pathway. Based on substrate specificity, SHP2 has a dual role in PI3K/Akt and JAK/STAT pathways.
Targeting SHP2 inhibition holds promise for combating immune checkpoint inhibitor (ICI) resistance by modulating critical signaling pathways. SHP2 inhibition could disrupt the MAPK, PI3K-AKT-mTOR, and JAK-STAT pathways, which are crucial for cancer cell survival and immune evasion. SHP2 inhibition has been suggested to attenuate MAPK pathway activation, impacting cell proliferation and survival. Moreover, SHP2 inhibition may hinder PI3K-AKT-mTOR signaling, curbing cancer cell growth and proliferation. Additionally, SHP2 inhibition has the potential to impact JAK-STAT signaling, which is critical for immune evasion mechanisms and tumor-mediated immunosuppression. These strategies targeting SHP2’s role in these signaling pathways could provide innovative approaches to enhance the efficacy of immune checkpoint blockade against resistant cancer cells.
RAS/ERK/SOS1
In addition to acting as an adaptor protein (activates RAS-RAF-ERK signaling by bridging upstream and downstream signals), SHP2 can also dephosphorylate downstream signaling molecules, activating downstream effectors in biological processes, including cell proliferation, migration, metabolism, and differentiation
[5]. Following growth factor stimulation, SHP2 is recruited by RTKs (SH2 domains link to the RTKs) and can function as an adaptor protein by attaching to phosphotyrosine-binding substrates like GAB1/2, GRB2, IRS1, FRS2, and Shc resulting in ERK activation
[27].
Grb2/SOS complex could be recruited to the tyrosine phosphorylation sites on SHP2 (Tyr542 and Tyr580), and RAS is activated in an SOS-dependent manner, leading to downstream stimulation of Raf1, MEK1/2 (intermediate kinases), and ERK1/2 (Raf1 downstream target). As a result, stimulated ERK1/2 induces the activity of several effectors, including transcription factors, which directly contribute to the proliferation and differentiation of cells
[43][44]. Through the activation of Src family kinases, SHP2 could indirectly activate RAS. Recruitment of SHP2 to Gab1 leads to dephosphorylation of paxillin and Cbp/PAG and the disconnection of Csk (a negative regulator of paxillin and src) and Src, and finally, results in src activation
[45].
PI3K/AKT
The crucial signaling system known as PI3K/AKT controls biological and physiological responses such as cell growth, survival, homeostasis, and metabolism
[44][46]. After activating EGFR, it recruits Gab1 through SHP2. Then, phosphorylated tyrosine Gab1 attracts PI3K and encourages the synthesis of PIP3 locally. Next, PIP3 can translocate Gab1 to the plasma membrane close to EGFR, which causes Gab1 to become phosphorylated and activates PI3K. Activated PI3K mediates the conversion of PI (4,5)P2 to PI (3,4,5)P3, which then employs AKT and PDK1, leading to cell survival and growth
[47]. Moreover, SHP2 can down-regulate PI3K activation by dephosphorylating Gab1
[48] and activate IRS1 through its phosphorylation
[27]. Thus, it seems that SHP2 has opposing functions in regulating the PI3K-AKT-mTOR pathway, depending on the stimuli.
JAK/STAT
STAT proteins, a family of transcription factors, are crucial for several biological processes, including cell growth, cell survival, differentiation, anti-apoptosis and cell motility
[49]. Based on substrate specificity, SHP2 has positive and negative functions in the JAK/STAT pathway. JAK2 activity and STAT5 phosphorylation are both reduced in SHP2-inactivated cells. SHP2 can dephosphorylate JAK and block the interaction of JAK with SOCS, reactivating the STAT signaling pathway
[27][50]. As a negative effect, the overexpression of SHP2 enhances STAT5’s dephosphorylation level in response to IL-3 stimulation, inhibiting STAT5 activity
[51][52]. The gp130 receptor typically transmits the JAK-STAT signaling that IL-6 generates, and mice lacking SHP2 showed prolonged activation of STAT3 by the gp130 receptor, suggesting a negative function of SHP2
[53]. The IL-6/IL-6R/gp130 complex recruits SHP2 and binds to the cytoplasmic tyrosine tail of gp130 to modulate downstream pathways, including dephosphorylation of STAT3 to negatively control the JAK-STAT3 signaling pathway and activating STAT3. This leads to the synthesis of PRL-3, which ultimately suppresses the activation of SHP2
[54].
7. SHP2 Inhibitors and Their Clinical Development in Oncology
Gain of function mutations of SHP2 plays a key role in the development and advancement of tumors and cancer by affecting cells directly
[55]. On the other hand, SHP2 plays a crucial role in numerous tumor-promotive and immune-suppressive signaling pathways within TME, cancer, and immune cells. Therefore, the inhibition of SHP2 can serve as a therapeutic approach.
7.1. SHP2 Inhibitors
SHP2 inhibitors generally include allosteric and orthosteric inhibitors
[56]. Orthosteric products bind at the active site, competing with the natural substrate or ligand and block the site from activation
[27][56]. Natural products like Tautomycetin (TTN) have shown promise as immunosuppressive and antitumor agents by selectively inhibiting SHP2 and reducing SHP2-dependent signaling. Suppressing SHP2 phosphatase with TTN may reduce SHP2-dependent signaling by reducing ERK1/2 activation
[56]. Other orthosteric inhibitors, such as cefsulodins, phenylhydrazonopyrazolone sulfonate derivatives, quinoline hydrazine derivatives, salicylic acid derivatives, diterpenoid quinone derivatives, and oxindole derivatives, have been studied for their potential inhibitory effects on SHP2
[56][57][58][59][60][61][62].
Conversely, allosteric inhibitors target sites other than the active site and change the binding site conformation. They have demonstrated advantages in selectivity and bioavailability compared to orthosteric inhibitors
[27]. Four allosteric sites exist, including the “tunnel” at the C-SH2/PTP domain interface. SHP099, SHP389, SHP394, SHP836, BBP-398, and TNO155 stabilize SHP2’s closed auto-inhibited state by attaching to the tunnel-like pocket; the “latch” at the N-SH2/PTP domain interface, such as SHP244 and its derivatives, SHP844 and SHP504; the “groove” at the N-SH2/PTP domain interface, which was on the other side of the tunnel and nonconserved cysteine residue 333 (Cys333) site, which is located in the PTP Domain
[63][64][65]. It should be noted that no allosteric inhibitors of SHP2 targeting the Groove-like site have been found
[27]. Overall, both orthosteric and allosteric inhibitors offer promising avenues for the development of targeted therapies against diseases involving SHP2-dependent signaling pathways.
7.2. SHP2 Inhibitors in the Clinic
While SHP2 inhibition as monotherapy has shown single-agent activity in solid tumors, a combinatorial approach synergizing known relevant pathways could overcome or delay adaptive resistance in tumors with certain oncogenic drivers. There are 11 small-molecule SHP2 inhibitors in mono- or combination clinical studies being evaluated for the treatment of cancer patients.
8. Overcoming Drug Resistance in Solid Tumors: SHP2 Inhibitors in Combination with Immune Checkpoint Inhibitors
Recent advancements in cancer research have focused on addressing drug resistance. To combat this challenge, researchers have explored innovative treatment approaches, and one promising strategy involves using SHP2 inhibitors in combination with immune checkpoint inhibitors such as PD-1, RTK, MEK, ERK, and ALK inhibitors. Activation of alternative receptor tyrosine kinases (RTKs) and downstream signaling cascades, such as the PI3K/AKT pathway, has been implicated in immune checkpoint therapy resistance
[66][67][68]. SHP2, as a downstream effector of multiple RTKs, plays a crucial role in these signaling networks. SHP2 plays a significant role in drug resistance mechanisms, making it an attractive therapeutic target
[27]. The deregulation of SHP2 is a frequent resistance mechanism in targeted treatments since it may operate as an oncogenic factor or tumor suppressor in various malignancies. Moreover, SHP2 regulates the suppression of immunological checkpoints, inhibiting patients’ antitumor immune responses, and making it an attractive therapeutic target
[66]. In general, many advanced tumors have more than one altered pathway, and combination strategies matched to the alterations present are associated with better outcomes
[69][70][71]. With SHP2, combination therapy has shown greater success than monotherapy in preclinical studies, overcoming drug resistance and issues arising from monotherapy
[72]. SHP2 inhibitors have demonstrated potential in addressing resistance to kinase inhibitors and PD-1 blockade
[27]. Resistance to TKIs can arise from various factors, including genetic alterations, gene amplification, and protein expression changes
[73]. TKIs can impair the function of ATP-binding cassette (ABC) transporters, making them useful in combination treatments. MDR-related ABC transporters regulate intracellular concentrations of small-molecule inhibitors. Combining SHP2 inhibitors with other protein inhibitors can overcome drug resistance
[27]. Inhibiting SHP2 provides a novel means of overcoming resistance mechanisms in cancer therapy, opening new possibilities for more effective and personalized treatments
[74].
SHP2 inhibitors can be logically combined with immunotherapies like checkpoint inhibitors due to their ability to affect anti-tumor activity through both tumor intrinsic and immune-mediated mechanisms
[74]. By combining SHP2 inhibitors with immune checkpoint inhibitors, researchers hope to achieve synergistic effects, overcome drug resistance, and improve treatment outcomes for patients with thoracic malignancies. Promising clinical combinations include SHP2 inhibitors with immune checkpoint inhibitors such as PD-1, RTK, MEK, ERK, and ALK inhibitors.
 +1 credit
+1 credit