Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Horváth, E.; Sólyom, �.; Székely, J.; Nagy, E.E.; Popoviciu, H. Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/51850 (accessed on 12 June 2026).
Horváth E, Sólyom �, Székely J, Nagy EE, Popoviciu H. Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/51850. Accessed June 12, 2026.
Horváth, Emőke, Árpád Sólyom, János Székely, Előd Ernő Nagy, Horațiu Popoviciu. "Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis" Encyclopedia, https://encyclopedia.pub/entry/51850 (accessed June 12, 2026).
Horváth, E., Sólyom, �., Székely, J., Nagy, E.E., & Popoviciu, H. (2023, November 21). Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. In Encyclopedia. https://encyclopedia.pub/entry/51850
Horváth, Emőke, et al. "Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis." Encyclopedia. Web. 21 November, 2023.
Copy Citation
Osteoarthritis (OA) is a complex disease of whole joints with progressive cartilage matrix degradation and chondrocyte transformation. The inflammatory features of OA are reflected in increased synovial levels of IL-1β, IL-6 and VEGF, higher levels of TLR-4 binding plasma proteins and increased expression of IL-15, IL-18, IL-10 and Cox2, in cartilage. Chondrocytes in OA undergo hypertrophic and senescent transition; in these states, the expression of Sox-9, Acan and Col2a1 is suppressed, whereas the expression of RunX2, HIF-2α and MMP-13 is significantly increased.
chondrocyte
inflammation
hypertrophy
senescence
metabolic
1. Introduction
Osteoarthritis (OA) is a complex disease that can affect any joint and involves a series of degenerative, inflammatory and remodeling processes. The early events in osteoarthritis are likely to involve the synovial membrane, which is well-vascularized, has mechanoreceptors and is therefore sensitive to systemic effects. In parallel or in rapid succession, abnormal mechanical loading and other factors may also result in chondrocyte phenotype switching and matrix remodeling; during the course of the disease, all parts of the joint, the articular cartilage, subchondral bone, synovial tissue and meniscus, interact in the development of pathological changes, including cartilage degradation, osteophyte formation, subchondral sclerosis and synovial hyperplasia [1]. The central change is cartilage degradation, which involves both cellular and extracellular phenomena. Inefficient repair contributes to the progression of cartilage deterioration. Chondrogenic progenitor cells are overrepresented in osteoarthritic cartilage compared to the normal joint. In early OA, mobile cell clusters form near the erosions and fissures, showing both anabolic and catabolic phenotypes (Notch-1, Stro-1, and VCAM-1). In advanced disease, migrating islands are distributed throughout the cartilage and can even pass through the fractured tidemark as shuttles between cartilage and subchondral bone. Their putative reparative role during disease progression is not fully understood, and the functional characterization of their subtypes is required to exploit their therapeutic potential [2]. In the course of OA, chondrocytes, the only cells of cartilage, are exposed to non-physiological stresses and biochemical stimuli from matrix degradation products and pro-inflammatory mediators [3]. These cells often undergo phenotypic changes with a shift towards senescence or apoptosis. Chondrocytes, together with synovial fibroblasts and macrophages and subchondral osteoclasts also undergo catabolic reprogramming, leading to matrix degradation and defective remodeling with altered collagen network and microarchitecture [4]. This functional transition is reflected in the presence of two closely related and histologically, immunologically and genetically detectable altered phenotypes, characterized by hypertrophy and senescence, which share several features and often coexist in a stressed environment due to abnormal biomechanical and biochemical challenges [5][6].
2. Evidence for the Presence of Low-Grade Inflammation in Osteoarthritis
The inflammation seen in OA differs from that characteristic of rheumatoid arthritis or other autoimmune arthritides. It is a chronic process, of low intensity compared to the aforementioned conditions, and is driven primarily by the innate immune system [7]. There are fewer macrophages and T-cells in OA than in joint tissue affected by rheumatoid arthritis [7]. All parts of the joint are affected: the synovial membrane and synovial fluid, the superficial and deep layers of cartilage, and the subchondral bone. Synovitis is responsible for some of the characteristic clinical symptoms: stiffness, warmth, pain, edema and joint effusion.
Sohn et al. identified 108 proteins of different classes (plasma proteins, serine protease inhibitors, markers of cartilage turnover and inflammatory cytokines) with altered levels in OA synovial fluid [8]. Despite the anatomical compartmentalization of the joint, inflammatory signaling in OA is not restricted to the synovial membrane, but is also characteristic of cartilage and subchondral bone. Disease-associated molecular patterns, such as calcium crystals, extracellular matrix degradation products, HMGB proteins, HSPs and the activation of the complement pathway, can lead to Nod-like receptor, TLR and RAGE signaling and maintain low-grade inflammation [9][10]. Interestingly, several plasma proteins such as Gc-globulin, α1-microglobulin and α2-macroglobulin can enhance pro-inflammatory cytokine production by macrophage cultures via the TLR-4 receptor [8]. Another study comparing synovial protein patterns in early and advanced OA and controls found increased levels of albumin, fibrinogen, alpha1-microglobulin/bikunin precursor, α2-macroglobulin, haptoglobin, ceruloplasmin and other acute phase proteins, and decreased levels of cystatin A and aggrecan 1 [11]. Among the inflammatory mediators, levels of IL-1β and VEGF have been found to be higher in osteoarthritic than in normal sera, while IL-6 can reach 190-fold higher concentrations in OA synovial fluid than in serum [8][12]. CXCL1 can promote IL-6 synthesis in synovial fibroblasts [13]. IL-1β, IL-6, TNFα and VEGF were all significantly elevated in the sera of 55 OA patients with different disease stages compared to controls [14].
In general, animal models support the presence of pro-inflammatory factors in OA, but in some cases there is still some controversy. Experimental OA reflects both primary and secondar, post-traumatic human disease, and includes spontaneous (naturally occurring and due to genetic background), induced (provoked surgically or chemically) and non-invasive murine, canine and lapine models applying a transarticular impact [15]. Meniscal ligament surgery in animals is sufficient to induce NF-kB activation, which is potentiated by IL-1β administration and reflected in high IL-6, MCP-1, MMP-13 and ADAMTS4 synthesis [16]. However, there are OA models in which the classical pathogenic role of IL-1 remains questionable. van Daelen et al. found that IL-1α and IL-1β are not involved in synovial inflammation and cartilage destruction in collagenase-initiated OA [17]. In meniscectomized IL-1β/NLRP3 double knockout mice induced with hydroxyapatite crystals, the severity and extent of cartilage lesions were even exacerbated compared to their meniscectomized counterparts [18].
In humans, in contrast to the variations of the aforementioned cytokines, changes in osteoarthritic plasma NF-kB and TNF-α levels may be moderate or even lower than in the healthy population [12]. A polyacrilamide gel–liquid chromatography–tandem mass spectrometry proteomics approach identified the presence of several factors and subunits of NF-kB in synovial fluid, such as NF-kB-repressing factor, NF-kB inhibitor-like protein 1, and NF-kB p100 subunit, which are normally found only in the intracellular space [8]. Stannus et al. performed a follow-up study of randomly enrolled subjects with a mean age of 63 years and measured baseline IL-6 and TNF-α levels compared with knee cartilage volume at baseline and after 3 years. The quartiles of IL-6 and TNF-α were associated with medial tibio-femoral joint space narrowing; the baseline IL-6 levels and change in IL-6 levels predicted the loss of medial and lateral cartilage volume [19]. In the study by Barker et al., TNF-α was lower in mild OA but not different in advanced OA than in controls, whereas IL-10 and the IL-10/TNF-α ratio were significantly lower in those with primary unilateral anterior cruciate ligament surgery and those who underwent total joint replacement [20]. In another human study, plasma leptin levels and synovial IL-18 were associated with Kellgren–Lawrence score [21]. In a small study group, Waszczykowski et al. observed increased serum levels of IL-18 and IL-20 compared to controls, with a significant correlation between IL-18 and MMP-3 levels [22]. IL-17 deficiency improved pain and cartilage degradation in IL-1Rα/IL-17 double knock-out mice with chemically (MIA)-induced OA [23]. The characteristic inflammatory changes seen in the synovial fluid and the bloodstream are represented in Figure 1.
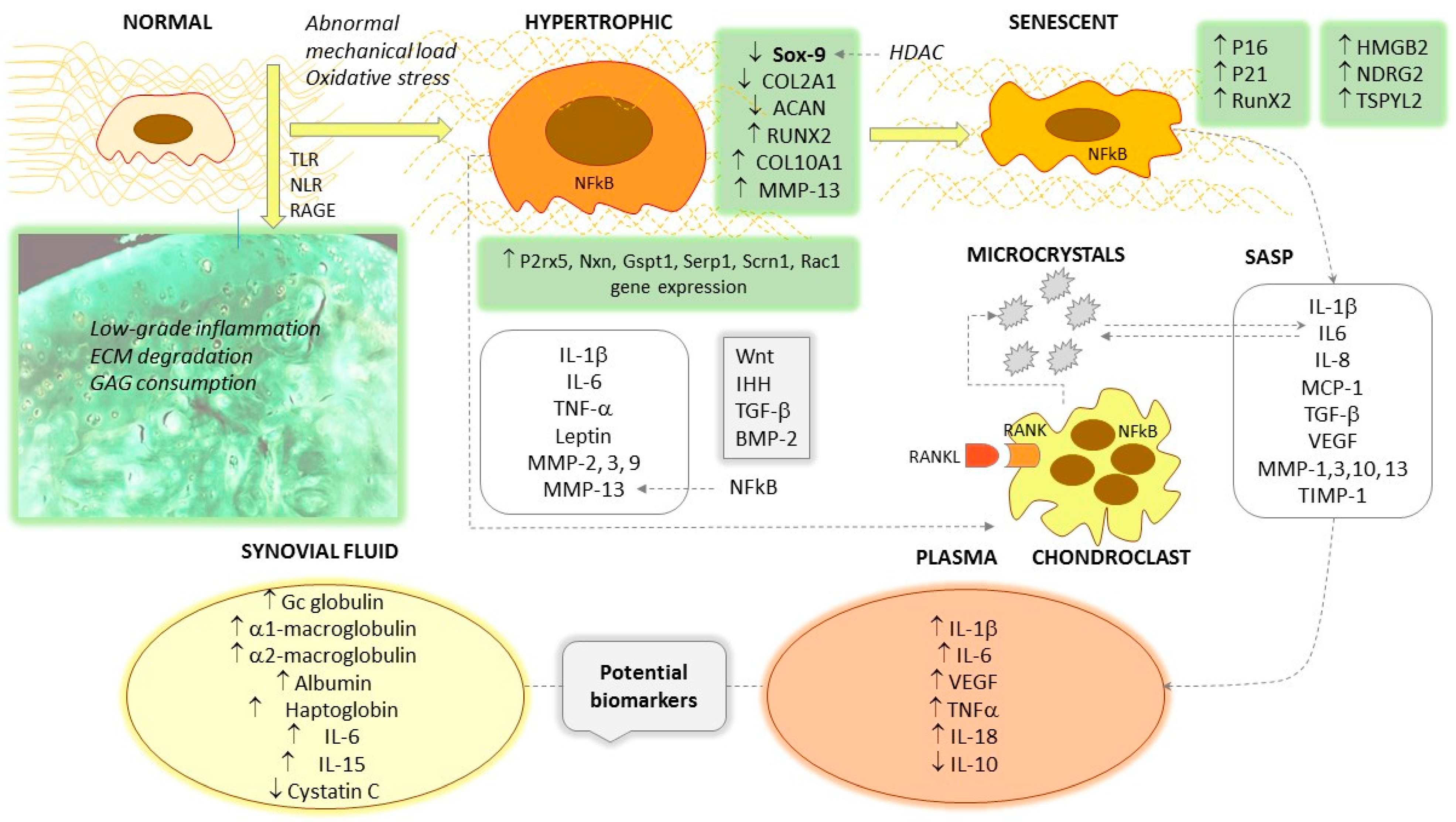
Figure 1. Hallmarks of the hypertrophic and senescent transformation of chondrocytes. ↑ symbolizes increased levels/stimulation; ↓ means decreased levels/inhibition.
Circulating cytokines can have many cellular sources, so there may be significant interference and high biological variability in their serum or plasma levels. They can originate from synovial macrophages and fibroblasts, subchondral osteoblasts, osteoclasts and chondrocytes, but also from other organs, with additive, synergistic or antagonistic effects on chondrocyte function. It is not easy to quantify locally produced and delivered samples, and powerful morphometric and imaging techniques are required for such tasks.
In terms of histological assessment, the expression of IL-1β, IL-6, TNF-α and VEGF proteins in the synovial membrane was significantly higher in moderate and advanced OA than in mild OA [14]. IL-6 appears to play a role in perpetuating inflammation, as it induces the deposition of hydroxyapatite crystals in human cartilage explants, providing a feedback loop for its synthesis [24]. Qu et al. found higher IL-6 and MMP-9 gene and protein expression in joint replacement surgery or arthroscopy specimens from OA patients compared to healthy controls [25]. Osteoarthritic synovial samples and LPS-induced synoviocytes showed upregulation of the nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3) inflammasome, in parallel with the redox homeostasis transcription factor Nrf2 and heme oxygenase-1 [26]. IL-1β and IL-18 were also overexpressed and decreased when Nrf2, but not NLRP3, was knocked down [26]. Warner et al. found that IL-15Rα was expressed in osteoarthritic cartilage samples, and IL-15 induced MMP-1 and MMP-3 synthesis in cultured cartilage explants. Genetic polymorphisms have been associated with the risk of symptomatic versus asymptomatic OA, and with the risk of neuropathic pain-like symptoms after total joint replacement surgery [27]. IL-15 was elevated in synovial fluid and synovial membrane cells obtained by arthroscopy in patients with early knee OA [28]. Counterregulatory mechanisms are also amplified, as high levels of IL-10 mRNA and protein have been detected in high-intensity cartilage lesions [29].
IL-1β and IL-6 trigger the cyclooxygenase pathway, and selective Cox-2 inhibition has raised high hopes as a candidate for disease-modifying drugs. Indeed, there is some experimental evidence that in chemically induced animal OA, meloxicam, a preferential Cox-2 inhibitor, has beneficial effects in inhibiting collagen type II degradation and cartilage destruction [30][31]. One possible trigger of the cyclooxygenase pathway is leptin: a concentration of 10 μg/mL increased the expression of Cox-2 and IL-6 as well as MMP-1, -3, -13, whereas suppressor of cytokine signaling-3 (SOCS-3) was able to stop these effects [32]. Serum leptin levels are elevated in obese OA patients, especially those with a high body-mass index (BMI), compared to non-OA, non-obese, and osteoarthritic individuals with low BMI, and correlate with the radiographic stage of the disease [33]. Moreover, 10 μg/mL leptin triggering alone, or with simultaneously administered IL-1β (10 pg/mL), stimulated the expression of inducible nitric oxide synthase, PGE2, IL-6 and IL-8 in femoral condyle and tibial plateau cartilage specimens obtained in the event of joint replacement [34].
3. Hypertrophic Differentiation and Chondrocyte Phenotype
Chondrocyte hypertrophy is a key feature of endochondral ossification, cartilage and bone development [35], and in the early stages of OA, dedifferentiated chondrocytes resemble their precursors in terms of proliferative and synthetic capacity. Hypertrophy represents terminal chondrocyte differentiation. Hypertrophy-like changes characterize both human and experimental OA, but do not involve all cells of the cartilage in a synchronized manner [36]. It has been proposed that hypertrophic differentiation of healthy articular chondrocytes occurs, at least in part, as a result of water entry through aquaporin channels [37]. The cells show increased organelle synthesis [35], and soluble compounds, and also the organic, stable microenvironment, interact with the differentiation process. In cell culture monolayers, regardless of their origin, chondrocytes lose their usual morphology and reorganize their cytoskeleton into an intensive production of thick actin fibers. Furthermore, the nuclear localization of myocardin-related transcription factor (MRTF) and increased expression of type I collagen (Col1) and tenascin C (Tnc) genes were observed, which was associated with the downregulation of Sox-9 gene expression [38].
Once differentiated during endochondral ossification, hypertrophic chondrocytes synthesize several metalloproteinases such as MMP-13, the gelatinases MMP-2 and MMP-9, and the stromelysins MMP-3 and MMP-10 [39]. In OA, hypertrophic chondrocytes indirectly mediate matrix remodeling by controlling chondroclasts, the inhabitants of the hyaline cartilage erosion zone [35]. These cells share morphological and transcriptomic features with osteoclasts, but are distinct in some features, with higher expression of the P2rx5, Nxn, Gspt1, Serp1, Scrn1 and Rac1 genes, but lower levels of ACP5 (tartrate-resistant acid phosphatase) [40]. Hypertrophic cartilage differs from normal cartilage in terms of the organization of both the cellular and extracellular matrix architecture. The superficial layer is disorganized, with focal matrix condensations, erosion and frequent matrix fibril ruptures. A characteristic glucosaminoglycan depletion can be seen in the deeper layers, which can be detected by safranin O staining [37]. In healthy cartilage, chondrocytes are homogeneously distributed, flattened in the superficial layer but round in the deep region. In contrast, osteoarthritic cartilage shows aggregated islands of chondrocytes of variable shape with the loss of characteristic organization and polarity. Experience gained in bioengineering experiments has shown that it is difficult to obtain a stable articular cartilage construct because of the shift to the so-called transient phenotype and hypertrophy instead of a stable permanent structure [37]. In the course of mesenchymal stem cell differentiation, several groups of signaling molecules dominate; BMP2,4,6, Indian hedgehog (IHH), parathormone-related peptide (PTHrP) signaling and the presence of angiogenic VEGF and pro-inflammatory IL-1β reflect commitment to the transient phenotype [37]. This transient cartilage contains high levels of collagen-I (COL-I), collagen-X (COL-X) and RUNX2, which confer a bone-like structure, and its final differentiation does indeed result in bone. In comparison, normal healthy articular chondrocytes characteristically express Sox-9 and COL-II, and GDF-5, autotaxin (ATX) and Chordin in their environment, induced mainly by the Wnt agonists Wnt4 and Wnt9a [41]. The phenotype, signaling, and secretory features of hypertrophic chondrocytes are represented in Figure 1.
The question that arises is whether chondrocyte hypertrophy is an obligatory feature of osteoarthritis. It appears that experimental hypertrophy is not always associated with OA, but a variable number of hypertrophic chondrocytes can be found in osteoarthritic cartilage.
Catheline et al. used the chondrocyte-specific overexpression of the transcription factor RunX2 with a Rosa26Runx2 allele, combined with inducible transgenes for COL2A and ACAN, and found that despite the early onset of chondrocyte hypertrophy, RunX2 overexpression alone was not sufficient to trigger OA. However, in a model of knee joint destabilization and meniscal injury, they described progressive cartilage degeneration characterized by impaired OARSI scores, increased apoptosis and high MMP-13 expression [42].
At least four activated signaling systems converge at the level of RunX2, distinguishing the signalomic signatures of normal and hypertrophic chondrocytes: a. the Wnt/LRP5/6/Fzd/β-catenin, b. the IHH/Ptch1, c. the TGF-β/TGFR/SMAD2,3,4, and d. the BMP2,4/SMAD1,5,8 [43]. Wnt ligands may play a dual role in the regulation of RunX2; Wnt 3, 8, 9 bind to the Frizzled receptor and enhance the nuclear translocation of β-catenin, and consequently the expression of RunX2 [37][44], triggering hypertrophy, whereas the non-canonical agonist Wnt 5a initially activates but later suppresses it by inhibiting RunX2 [45]. Ferrao Blanco et al. reported interesting results on the hypertrophic phenotype of human articular chondrocytes induced by inflammatory cytokines or medium conditioned by defined macrophage subsets. They found that genes associated with hypertrophy (Col10a1 and RunX2) were downregulated by the mixture of cytokines IL-1β, TNF-α and IFN-γ in cartilage explants and chondrocytes packed in alginate, and only MMP-13 expression could be upregulated by these stimuli. NF-kB inhibition with SC-514 did not significantly alter the expression of Col10a1, RunX2 and MMP-13, nor did the addition of medium-conditioned TNF-α/IFN-γ producing macrophages. On the contrary, IL-4-synthesising reparative macrophages were able to upregulate COL10A1, RUNX2 and IHH in cartilage explants, which the authors considered to be more important than the intrinsic inflammatory signals [46]. The role of macrophages in OA is also supported by the finding that macrophage depletion in mice protects against collagenase-induced OA [47].
Due to abnormal mechanical loading, excessive oxidative stress and other factors, the normal chondrocyte in OA undergoes hypertrophic transformation. In addition to increased size, this phenotype is manifested by the overexpression of Sox-9, RunX2 and COL10A1 and the suppression of COL2A1 and ACAN. The Wnt, IHH, TGF-β and BMP-2 signal pathways are amplified, and pro-inflammatory cytokines and several metalloproteinases such as MMP-2, MMP-3, MMP-9 and MMP-13 appear in the secretory pattern. Osteoarthritic cartilage shows low-grade inflammation and the degradation of the extracellular matrix (collagen type II, aggrecan and glycosaminoglycan components). The accumulation of microcrystals favors the transformation, and they have a mutual triggering effect with IL-6. The final differentiation of hypertrophic chondrocytes results senescent cells, characterized by the overexpression of P16 and p21, increased RunX2 and the newly discovered HMGB2, NDRG2 and TSPYL2. The senescence-associated secretion pattern includes IL-1β, IL-6, IL-8, MCP-1, VEGF, MMP-1, MMP-3 and MMP-13. As elevated levels can be detected in the bloodstream, some of these can be considered potential biomarkers for OA. Some cytokines, such as IL-6 and IL-15, also show characteristic elevations in synovial fluid.
References
- Chen, D.; Shen, J.; Zhao, W.W.; Wang, T.Y.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044.
- Jiang, Y.Z.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 206–212.
- Aigner, T.; Soeder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of Disease: Role of chondrocytes in the pathogenesis of osteoarthritis—Structure, chaos and senescence. Nat. Clin. Pract. Rheumatol. 2007, 3, 391–399.
- Yoshioka, N.K.; Young, G.M.; Khajuria, D.K.; Karuppagounder, V.; Pinamont, W.J.; Fanburg-Smith, J.C.; Abraham, T.; Elbarbary, R.A.; Kamal, F. Structural changes in the collagen network of joint tissues in late stages of murine OA. Sci. Rep. 2022, 12, 9159.
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358.
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218.
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592.
- Sohn, D.H.; Sokolove, J.; Sharpe, O.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Lindstrom, T.M.; Hwang, I.; Boyer, K.A.; Andriacchi, T.P.; et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res. Ther. 2012, 14, R7.
- Terkawi, M.A.; Ebata, T.; Yokota, S.; Takahashi, D.; Endo, T.; Matsumae, G.; Shimizu, T.; Kadoya, K.; Iwasaki, N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines 2022, 10, 1109.
- van den Bosch, M.H.J. Inflammation in osteoarthritis: Is it time to dampen the alarm(in) in this debilitating disease? Clin. Exp. Immunol. 2019, 195, 153–166.
- Gobezie, R.; Kho, A.; Krastins, B.; Sarracino, D.A.; Thornhill, T.S.; Chase, M.; Millett, P.J.; Lee, D.M. High abundance synovial fluid proteome: Distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 2007, 9, R36.
- Krenytska, D.; Strubchevska, K.; Kozyk, M.; Vovk, T.; Halenova, T.; Kot, L.; Raksha, N.; Savchuk, O.; Falalyeyeva, T.; Tsyryuk, O.; et al. Circulating levels of inflammatory cytokines and angiogenesis-related growth factors in patients with osteoarthritis after COVID-19. Front. Med. 2023, 10, 1168487.
- Hou, S.M.; Chen, P.C.; Lin, C.M.; Fang, M.L.; Chi, M.C.; Liu, J.F. CXCL1 contributes to IL-6 expression in osteoarthritis and rheumatoid arthritis synovial fibroblasts by CXCR2, c-Raf, MAPK, and AP-1 pathway. Arthritis Res. Ther. 2020, 22, 251.
- Wang, Z.W.; Chen, L.; Hao, X.R.; Qu, Z.A.; Huang, S.B.; Ma, X.J.; Wang, J.C.; Wang, W.M. Elevated levels of interleukin-1 beta, interleukin-6, tumor necrosis factor-alpha and vascular endothelial growth factor in patients with knee articular cartilage injury. World J. Clin. Cases 2019, 7, 1262–1269.
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016, 11, 19.
- Arra, M.; Swarnkar, G.; Alippe, Y.; Mbalaviele, G.; Abu-Amer, Y. I kappa B-zeta signaling promotes chondrocyte inflammatory phenotype, senescence, and erosive joint pathology. Bone Res. 2022, 10, 12.
- Van Dalen, S.C.M.; Blom, A.B.; Sloetjes, A.W.; Helsen, M.M.A.; Roth, J.; Vogl, T.; de Loo, F.A.J.; Koenders, M.I.; Van der Kraan, P.M.; Van den Berg, W.B.; et al. Interleukin-1 is not involved in synovial inflammation and cartilage destruction in collagenase-induced osteoarthritis. Osteoarthr. Cartil. 2017, 25, 385–396.
- Nasi, S.; Ea, H.K.; So, A.; Busso, N. Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1 alpha and-1 beta, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of osteoarthritis. Front. Pharmacol. 2017, 8, 282.
- Stannus, O.; Jones, G.; Cicuttini, F.M.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 2010, 18, 1441–1447.
- Barker, T.; Rogers, V.E.; Henriksen, V.T.; Trawick, R.H.; Momberger, N.G.; Lynn Rasmussen, G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci. Rep. 2021, 11, 1812.
- Panina, S.B.; Krolevets, I.V.; Milyutina, N.P.; Sagakyants, A.B.; Kornienko, I.V.; Ananyan, A.A.; Zabrodin, M.A.; Plotnikov, A.A.; Vnukov, V.V. Circulating levels of proinflammatory mediators as potential biomarkers of post-traumatic knee osteoarthritis development. J. Orthop. Traumatol. 2017, 18, 349–357.
- Waszczykowski, M.; Fabis-Strobin, A.; Bednarski, I.; Narbutt, J.; Fabis, J. Serum and synovial fluid concentrations of interleukin-18 and interleukin-20 in patients with osteoarthritis of the knee and their correlation with other markers of inflammation and turnover of joint cartilage. Arch. Med. Sci. 2022, 18, 448–458.
- Na, H.S.; Park, J.S.; Cho, K.H.; Kwon, J.Y.; Choi, J.; Jhun, J.; Kim, S.J.; Park, S.H.; Cho, M.L. Interleukin-1-Interleukin-17 Signaling Axis Induces Cartilage Destruction and Promotes Experimental Osteoarthritis. Front. Immunol. 2020, 11, 730.
- Nasi, S.; So, A.; Combes, C.; Daudon, M.; Busso, N. Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann. Rheum. Dis. 2016, 75, 1372–1379.
- Qu, X.Q.; Wang, W.J.; Tang, S.S.; Liu, Y.; Wang, J.L. Correlation between interleukin-6 expression in articular cartilage bone and osteoarthritis. Genet. Mol. Res. 2015, 14, 14189–14195.
- Jhang, J.J.; Yen, G.C. The role of Nrf2 in NLRP3 inflammasome activation. Cell Mol. Immunol. 2017, 14, 1011–1012.
- Warner, S.C.; Nair, A.; Marpadga, R.; Chubinskaya, S.; Doherty, M.; Valdes, A.M.; Scanzello, C.R. IL-15 and IL15RA in Osteoarthritis: Association With Symptoms and Protease Production, but Not Structural Severity. Front. Immunol. 2020, 11, 1385.
- Scanzello, C.R.; Umoh, E.; Pessler, F.; Diaz-Torne, C.; Miles, T.; DiCarlo, E.; Potter, H.G.; Mand, L.; Marx, R.; Rodeo, S.; et al. Local cytokine profiles in knee osteoarthritis: Elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthr. Cartil. 2009, 17, 1040–1048.
- Iannone, F.; De Bari, C.; Dell’Accio, F.; Covelli, M.; Cantatore, F.P.; Patella, V.; Lo Bianco, G.; Lapadula, G. Interleukin-10 and interleukin-10 receptor in human osteoarthritic and healthy chondrocytes. Clin. Exp. Rheumatol. 2001, 19, 139–146.
- Nagy, E.; Vajda, E.; Vari, C.; Sipka, S.; Farr, A.M.; Horvath, E. Meloxicam ameliorates the cartilage and subchondral bone deterioration in monoiodoacetate-induced rat osteoarthritis. PeerJ 2017, 5, e3185.
- Csifo, E.; Nagy, E.E.; Horvath, E.; Farr, A.; Muntean, D.L. Mid-term effects of Meloxicam on collagen type II degradation in a rat osteoarthritis model induced by iodoacetate. Farmacia 2015, 63, 556–560.
- Koskinen-Kolasa, A.; Vuolteenaho, K.; Korhonen, R.; Moilanen, T.; Moilanen, E. Catabolic and proinflammatory effects of leptin in chondrocytes are regulated by suppressor of cytokine signaling-3. Arthritis Res. Ther. 2016, 18, 215.
- Lambova, S.N.; Batsalova, T.; Moten, D.; Stoyanova, S.; Georgieva, E.; Belenska-Todorova, L.; Kolchakova, D.; Dzhambazov, B. Serum Leptin and Resistin Levels in Knee Osteoarthritis-Clinical and Radiologic Links: Towards Precise Definition of Metabolic Type Knee Osteoarthritis. Biomedicines 2021, 9, 1019.
- Vuolteenaho, K.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Päivärinta, U.; Moilanen, T.; Moilanen, E. Leptin Enhances Synthesis of Proinflammatory Mediators in Human Osteoarthritic Cartilage-Mediator Role of NO in Leptin-Induced PGE2, IL-6, and IL-8 Production. Mediat. Inflamm. 2009, 2009, 345838.
- Hallett, S.A.; Ono, W.; Ono, N. The hypertrophic chondrocyte: To be or not to be. Histol. Histopathol. 2021, 36, 1021–1036.
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232.
- Majumder, N.; Ghosh, S. Unfolding the Mystery Behind the Onset of Chondrocyte Hypertrophy during Chondrogenesis: Toward Designing Advanced Permanent Cartilage-mimetic Biomaterials. Adv. Funct. Mater. 2023, 33, 2300651.
- Parreno, J.; Raju, S.; Niaki, M.N.; Andrejevic, K.; Jiang, A.; Delve, E.; Kandel, R. Expression of type I collagen and tenascin C is regulated by actin polymerization through MRTF in dedifferentiated chondrocytes. Febs Lett. 2014, 588, 3677–3684.
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93.
- Khan, N.M.; Clifton, K.B.; Lorenzo, J.; Hansen, M.F.; Drissi, H. Comparative transcriptomic analysis identifies distinct molecular signatures and regulatory networks of chondroclasts and osteoclasts. Arthritis Res. Ther. 2020, 22, 168.
- Biswas, T.; Jaswal, A.P.; Yadav, U.S.; Bandyopadhyay, A. Simultaneous differentiation of articular and transient cartilage: WNT-BMP interplay and its therapeutic implication. Int. J. Dev. Biol. 2020, 64, 213–221.
- Catheline, S.E.; Hoak, D.; Chang, M.; Ketz, J.P.; Hilton, M.J.; Zuscik, M.J.; Jonason, J.H. Chondrocyte-Specific RUNX2 Overexpression Accelerates Post-traumatic Osteoarthritis Progression in Adult Mice. J. Bone Miner. Res. 2019, 34, 1676–1689.
- Chawla, S.; Mainardi, A.; Majumder, N.; Donges, L.; Kumar, B.; Occhetta, P.; Martin, I.; Egloff, C.; Ghosh, S.; Bandyopadhyay, A.; et al. Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies. Cells 2022, 11, 4034.
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18.
- Bradley, E.W.; Drissi, M.H. WNT5A Regulates Chondrocyte Differentiation through Differential Use of the CaN/NFAT and IKK/NF-kappa B Pathways. Mol. Endocrinol. 2010, 24, 1581–1593.
- Ferrao Blanco, M.N.; Bastiaansen-Jenniskens, Y.M.; Chambers, M.G.; Pitsillides, A.A.; Narcisi, R.; van Osch, G.J.V.M. Effect of Inflammatory Signaling on Human Articular Chondrocyte Hypertrophy: Potential Involvement of Tissue Repair Macrophages. Cartilage 2021, 13 (Suppl. S2), 168S–174S.
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The Role of Synovial Macrophages and Macrophage-Produced Mediators in Driving Inflammatory and Destructive Responses in Osteoarthritis. Arthritis Rheum. 2010, 62, 647–657.
More
Information
Subjects:
Rheumatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
706
Revisions:
2 times
(View History)
Update Date:
22 Nov 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No