Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gongqin Sun | -- | 2355 | 2023-11-08 15:05:19 | | | |
| 2 | Rita Xu | Meta information modification | 2355 | 2023-11-09 02:33:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chapdelaine, A.G.; Sun, G. Targeted Therapies for Triple Negative Breast Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/51302 (accessed on 05 July 2026).
Chapdelaine AG, Sun G. Targeted Therapies for Triple Negative Breast Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/51302. Accessed July 05, 2026.
Chapdelaine, Abygail G., Gongqin Sun. "Targeted Therapies for Triple Negative Breast Cancer" Encyclopedia, https://encyclopedia.pub/entry/51302 (accessed July 05, 2026).
Chapdelaine, A.G., & Sun, G. (2023, November 08). Targeted Therapies for Triple Negative Breast Cancer. In Encyclopedia. https://encyclopedia.pub/entry/51302
Chapdelaine, Abygail G. and Gongqin Sun. "Targeted Therapies for Triple Negative Breast Cancer." Encyclopedia. Web. 08 November, 2023.
Copy Citation
Triple negative breast cancer (TNBC) is a heterogeneous group of breast cancers characterized by their lack of estrogen receptors, progesterone receptors, and the HER2 receptor. They are more aggressive than other breast cancer subtypes, with a higher mean tumor size, higher tumor grade, the worst five-year overall survival, and the highest rates of recurrence and metastasis.
triple negative breast cancer
targeted therapy
multi-driver oncogenesis
1. Introduction
Breast cancer is the second leading cause of cancer-related deaths in American women [1][2]. About 15% of breast cancers [3][4][5] lack estrogen receptors (ER), progesterone receptors (PR), and HER2 [6][7]. Such cases are referred to as triple negative breast cancer (TNBC). TNBC shares ~75% overlap with basal-like breast cancer, and its cells tend to look like the epithelial cells of the outmost, basal layer of the breasts’ milk ducts. TNBC is more aggressive than other breast cancer subtypes [7][8][9], with a higher mean tumor size, higher tumor grade, the worst five-year overall survival, and the highest rates of recurrence and metastasis [10][11][12]. It is more common in premenopausal and African American women [13][14][15][16].
While ER+ and HER2+ breast cancer patients significantly benefit from targeted therapies blocking ER [17][18] and HER2 [12][19][20], a lack of hormonal targets in TNBC has left patients with minimal treatment options. As such, alternative targets need to be discovered and exploited for treating TNBC. Significant effort has been invested into finding such targets and developing targeted therapies for TNBC; however, they have yielded limited success, and no broadly effective targeted therapy has been approved for TNBC. Consequently, TNBC treatment still relies on chemotherapy, surgery, and radiation therapy [21][22]. While this approach has been successful in early-stage TNBCs, it is relatively ineffective in advanced-stage patients, reflected in the fact that metastatic TNBC has a 5-year survival rate of only 12% [23][24]. Finding targeted therapy treatments for TNBC patients is desperately needed, and the struggle in developing such an approach epitomizes the limited reach of targeted cancer therapies in general. In 2018, only 8.33% of all US cancer patients were genomically eligible for targeted therapies and only 4.9% benefited from the treatment [25].
2. Developing Combination Targeted Therapy for TNBC
As accumulating evidence supports the concept of multi-driver oncogenesis in TNBC, it becomes evident that drug combinations simultaneously blocking multiple drivers would be necessary to treat TNBC.
2.1. Multi-Driver Oncogenesis and Combination Targeted Therapy
As discussed above, there is strong evidence that most TNBC cancers contain multiple oncogenic drivers. The likely multi-driver nature of TNBC is not surprising, as most cancers are multi-driver cancers [26][27][28][29][30][31]. Cancer development is an evolutionary process of selecting cells with growth and survival advantages in a tumor micro-environment [26][32][33]. Multiple growth and survival drivers would confer such a selective advantage. A recent study [31] of 7664 tumors of 29 types revealed that a tumor carries four driver mutations on average, but the number varies widely (from 1 to >10) among cancer types. Frequent mutations and amplification in rPTKs, PI3K and MAPK pathways, and Src upregulation in TNBC likely activate multiple drivers [3]. The lack of success of monotherapy against a broad array of oncogenic drivers also supports the multi-driver nature of TNBC.
Identifying drug combinations for cancers mostly relies on empirical screening [34][35][36][37]. For example, Wali et al. [35] assessed 768 drug combinations between 128 drug candidates and six FDA-approved drugs on TNBC cells. Such studies can identify effective drug combinations; however, an insufficient number of combinations, an incomplete coverage of prospective drivers, and the absence of mechanistic considerations limit the potential of this approach. Some clinical trials also empirically formulate drug combinations [38] but lack a strategy for mechanism-based formulations. A 2017 analysis [39] of current drug combinations in clinical trials and patient-derived xenograft animal models revealed that most of the benefits of combination cancer therapies were due to different patient subgroups benefiting from different components of a combination rather than from synergy or additivity of the combination on individual patients. Truly harnessing the power of combination targeted therapy is dependent on identifying the oncogenic drivers and developing mechanism-based drug combinations to block all drivers in TNBC.
2.2. Current Pharmacological Models Are Not Suitable for Analyzing the Drug Response of Multi-Driver Cancers
Developing drug combinations that simultaneously block multiple drivers is a unique challenge for cancer. Modern drug discovery is largely focused on finding and optimizing drugs against individual targets. The current pharmacological analysis is based on a one-drug-one-target paradigm coded in several versions of the Hill equation [40]. It is often expressed as follows:
I = Imax × Dn/((IC50*)n + Dn))
In this equation, the inhibitory effect (I) is a function of maximal inhibition (Imax), drug concentration (D), half inhibitory concentration (IC50), and inhibitory slope or the Hill Co-efficient (n). The IC50 reflects the affinity between the drug and the target, and “n” measures the cooperativity in binding. When n is above 1, there is positive cooperativity, famously exemplified by O2 binding to hemoglobin. When n is below 1, it is assumed to be “negative cooperativity”, which remains a mechanistic enigma in modern pharmacology [41]. Because Hill equation-based pharmacology interprets drug response based on a one-drug-one-target paradigm, it is not adequate in characterizing the effects of a kinase-based targeted drug on multi-driver cancer cells, where one drug can exert its effects by inhibiting multiple targets [42][43].
Accumulating evidence indicates that cancer cell drug responses cannot be readily described by the pharmacological models represented by the Hill equation. Many cancer cells display unusually shallow response curves when treated by targeted drugs, especially those blocking the Akt/PI3K/mTOR pathway [41]. The shallow inhibition curves fall into the “negative cooperativity” category that has no ready mechanistic explanation [41]. Another report found that 28% of cancer drug responses are multiphasic [44]. A new paradigm for analyzing such complex responses was recently developed.
2.3. A Strategy for Mechanism-Based Targeted Drug Combination
Based on the analysis of shallow and multiphasic inhibition, a new mathematical model for analyzing the effects of targeted drugs on multi-driver cancers was recently developed. Shen et al. determined that shallow inhibition is biphasic in nature consisting of a potent target-specific inhibition and less potent off-target inhibition. This is a unique feature for the effects of targeted drugs on multi-driver cancer cells, as mono-driver cancer cells are killed by drugs in a monophasic manner [42][43][45]. To quantify the biphasic nature of shallow inhibition, they developed a biphasic model represented by the equation below.
I = F1 × [D]/([D] + Kd1) + F2 × [D]/([D] + Kd2)
In this model, the inhibition (I) by a drug has two phases: F1 and F2 as fractions of total cell viability (F1 + F2 = 100%), and each phase has its own binding affinity (Kd1 and Kd2). Curve-fitting shallow dose-response data to this equation yields F1, F2, Kd1, and Kd2. This analysis reveals the relative role a potential driver plays in the viability of a multi-driver cancer cell (F1) and the potency of the driver inhibition (Kd1). Thus, this analysis allowed the identification of drugs for each driver in a multi-driver cancer.
Different drug dose-response patterns by mono-driver and multi-cancer driver cancer cells are illustrated in Figure 1. In a mono-driver cancer cell (Figure 1A), a drug blocking the driver causes a mono-phasic dose-response pattern (Figure 1B). In a multi-driver cancer cell (Figure 1C), a drug blocking one driver would cause only a partial inhibition, such as curve 1 in Figure 1D. Sometimes, a drug may cause additional off-target inhibition, generating a biphasic curve as illustrated as curve 2 in Figure 1D. When inhibitors for different drivers are combined, each inhibitor is blocking its own target, and the combination would simultaneously block both drivers, leading to synergistic inhibition of cell viability, generating a monophasic like dose-response curve. This prediction has been confirmed in numerous colorectal cancer models [43][46] and TNBC cancer cells [42][47].
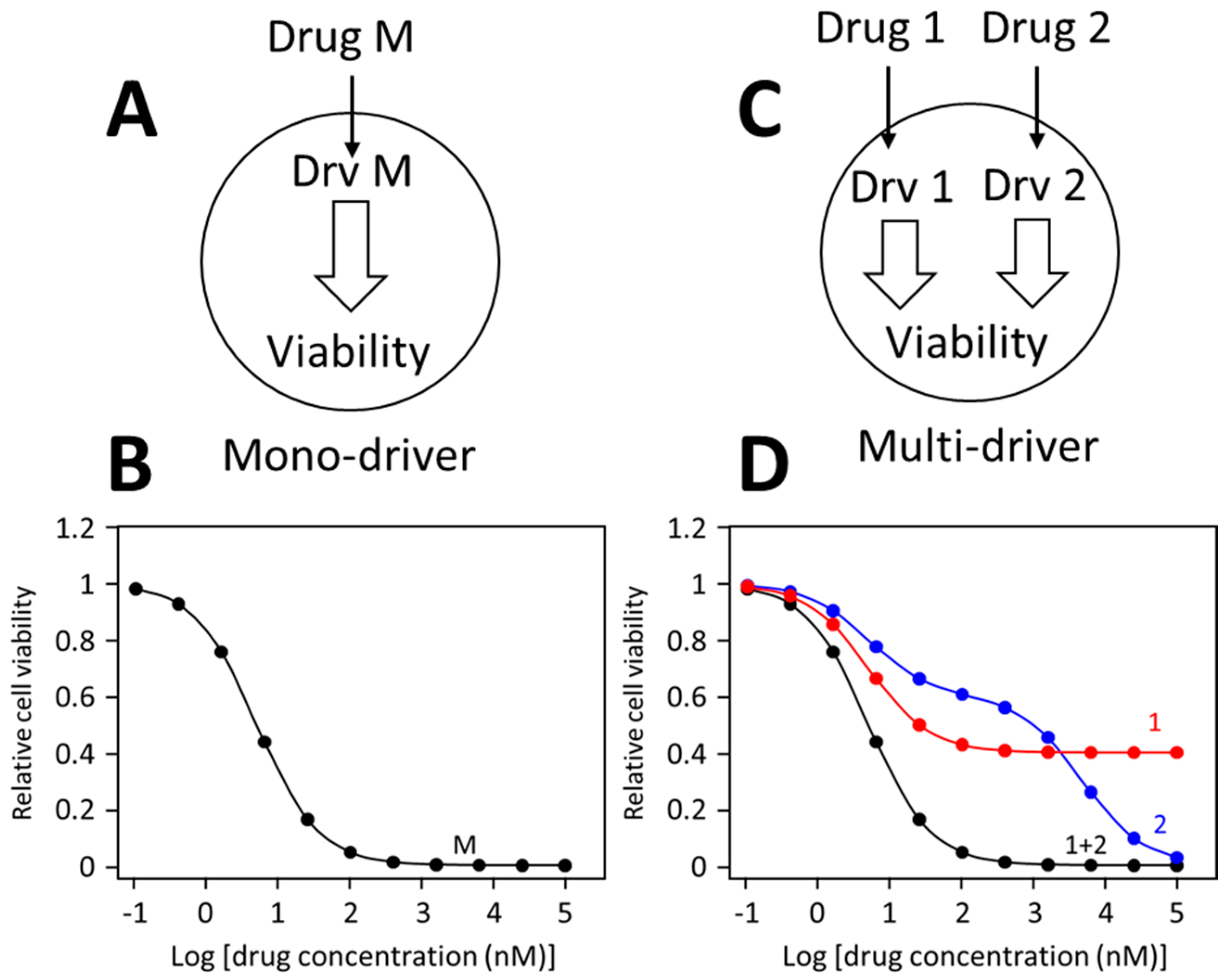
Figure 1. Dose-response curves of mono-driver and multi-driver cancer cells to targeted drugs. (A) Depiction of a mono-driver cell. (B) A monophasic dose-response curve by the mono-driver cancer cell to an inhibitor blocking its driver (Drv M). (C) Depiction of a multi-driver cancer cells with driver 1 (Drv 1) driver 2 (Drv 2). (D) Response of the multi-driver cancer cell to inhibitor for each driver individually or in combination. These dose-response curves illustrate idealized response patterns.
Applying this strategy to TNBC, highly potent drug combinations for the cell lines MDA-MB-231 and MDA-MB-468 were identified. Shen et al. determined that MDA-MB-231 contained two oncogenic drivers, Src and KRAS (due to a G13D mutation), and the cell proliferation can be partially inhibited by either the Src inhibitor dasatinib or Mek inhibitors trametinib or selumetinib. Each drug alone causes a shallow and biphasic inhibition of MDA-MB-231 cells and does not lethally inhibit MDA-MB-231; however, the combination of dasatinib and trametinib can lethally inhibit MDA-MB-231 cells with an IC50 of 8.2 nM. The combination also displays striking synergy. For example, the IC70 (drug concentration for 70% inhibition) is 25 nM for the combination, 12.6 μM for dasatinib, and above 20 μM for trametinib, resulting in a combination index (CI) of <0.003, and a dose reduction index (DRI) > 300 [48][49]. Thus, the combination is >300-fold more potent than dasatinib/trametinib without synergy.
The same approach also identified a potent drug combination for MDA-MB-468. It was demonstrated that MDA-MB-468 proliferation and survival is dependent on EGFR over-expression and the activated PI3K pathway due to low expression of PTEN [42]. The cells are partially sensitive to both lapatinib (an EGFR inhibitor) [50][51] and GSK690693 (an Akt inhibitor) [52][53], and are potently inhibited by the lapatinib/GSK690693 combination (IC50 = 22 nM). The drug combination is also strikingly synergistic, with a CI of 0.025 and DRI of 40 at 70% inhibition [42].
The combinations are also strikingly specific for MDA-MB-231 and MDA-MB-468. Dasatinib + trametinib is 1800-fold more potent to MDA-MB-231 than MDA-MB-468 (IC50 of 8.2 nM vs. 15 μM), and lapatinib + GSK690693 is 454-fold more potent to MDA-MB-468 than MDA-MB-231 (IC50 of 22 nM vs. 10 μM). The specificity indicates that the inhibition is mechanism-based. This strategy of developing combination targeted therapy is yet to be verified in animal models and in clinical settings, but it offers a strategy to identify potent, synergistic, and mechanism-based targeted drug combinations for multi-driver cancers, such as TNBC.
3. New and Emerging Targets and Treatments for TNBC
New targets and new targeting strategies are emerging from preclinical studies of TNBC. These include targeting metabolism, epigenetic regulation, and developing new methods to target proteins and enzymes. These strategies could provide new treatment options in the future for TNBC patients.
3.1. Targeting Metabolism
One emerging hallmark of cancer is reprogrammed energy metabolism [54], where cancer cells greatly increase glucose uptake and become more reliant on hyperactivated glycolysis and less reliant on decreased oxidative phosphorylation for energy production. As such, one area of potential targets are metabolic enzymes in glycolysis, oxidative phosphorylation, and lipid metabolism. The reprogrammed metabolism not only promotes cell proliferation but also contributes to drug resistance to chemotherapy. Thus, targeting metabolism has the potential both as a direct therapeutic approach and as a tool to counter drug resistance.
Multiple targets have been studied to manipulate glycolysis, such as inhibiting glucose transporters (e.g., small molecules WZB117 and resveratrol) or glycolysis (e.g., 2-deoxy-D-glucose [55] as a glucose analog, metformin inhibiting hexokinase, and 3-bromopyruvate inhibiting glyceraldehyde-3-phosphate dehydrogenase). Even though oxidative phosphorylation is reduced in cancer cells, it still plays an essential role in TNBC cells, which makes oxidative phosphorylation a potential therapeutic vulnerability [56]. IAC-10759 is a novel inhibitor of complex I of the mitochondrial electron transport chain [57]. It inhibits the growth of a broad range of patient-derived xenograft TNBC tumors [56], and it is being tested for its therapeutic efficacy against TNBC and other solid tumors in a clinical trial (NCT03291938).
Other metabolic functions may also provide therapeutic targets for TNBC. Recent evidence demonstrates that upregulation of essential lipogenic enzymes acetyl-CoA carboxylase-α and fatty acid synthase enhances the malignant behavior of TNBC [57][58][59][60]. Mitochondrial morphology and dynamics have also been associated with TNBC tumor growth and metastasis [58][61].
3.2. Epigenetic Therapy
Epigenetic regulation of gene activity is exerted by a number of mechanisms, such as DNA methylation, histone Lys acetylation and methylation, and several forms of non-coding RNAs [62]. With the development and progression of a tumor, there is a progressive loss of total DNA methylation, an increase of hypermethylated CpG islands, and an increased histone modification [63][64][65]. Hypermethylation of the CpG islands causes the inactivation of numerous tumor suppressor genes, including BRCA1 [66][67]. In TNBC, epigenetic modifications are known to play a crucial role in the epithelial-mesenchymal transition (EMT) and metastasis [68][69]. Numerous anti-cancer therapeutic targets in epigenetic modifications have emerged, and small molecule inhibitors for enzymes in maintaining DNA methylation and histone modifications are being actively studied as therapeutic approaches to counter oncogenic processes (reviewed in [70][71]). Epigenetic therapy could prove particularly attractive for TNBC because of the lack of alternative targeted therapies.
3.3. New Therapeutic Modalities
In addition to the traditional enzyme inhibitors and monoclonal antibodies that make up most current targeted therapeutics, other therapeutic modalities, such as small molecules disrupting protein-protein interaction and Proteolysis Targeting Chimeras (PROTACs) could provide new ways to manipulate oncogenic molecular processes. These new intervening modalities significantly expand the number of targets that can be manipulated chemically.
A large number of cellular processes are regulated or mediated by protein–protein interaction, and targeting protein–protein interaction has been recognized as a valuable approach to manipulate cancer cell biology [72]. For example, p53 is a tumor suppressor lost in most TNBC tumors due to mutation (84%) or other pathway inactivating events, such as gain of MDM2 (14%) [3]. Restoring p53 function could be a useful approach to inhibit cancer progression, but enzyme inhibitors or monoclonal antibodies are not useful for this purpose. Small molecules activating p53 or blocking its interactions with MDM2 are able to restore p53 pathway function and make cancers more sensitive to anti-mitotic drugs [73][74]. Of note is the 2-sulfonylpyrimidine compound PK11007, which alkylates p53 on specific residues and increases p53 thermal stability. PK11007 inhibits cell proliferation, induces apoptosis, and alters the expression of genes involved in cell death in TNBC cells [75].
Another approach for targeting the non-enzymatic functions of proteins in cancer cells is the usage of PROTACs [76][77][78]. PROTACs are heterobifunctional small molecules consisting of two ligands joined by a linker: one ligand binds a target protein while the other binds an E3 ubiquitin ligase. Simultaneous binding of the target protein and ligase by the PROTAC induces ubiquitylation of the target protein and its subsequent degradation by the ubiquitin–proteasome system [78]. This technology allows specific degradation of the target protein. A number of PROTACs are in clinical trials for cancer therapy [78].
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70.
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519.
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948.
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609.
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392.
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690.
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23 (Suppl. S6), vi7–vi12.
- Pal, S.K.; Childs, B.H.; Pegram, M. Triple negative breast cancer: Unmet medical needs. Breast Cancer Res. Treat. 2011, 125, 627–636.
- Schroeder, M.C.; Rastogi, P.; Geyer, C.E.; Miller, L.D.; Thomas, A. Early and Locally Advanced Metaplastic Breast Cancer: Presentation and Survival by Receptor Status in Surveillance, Epidemiology, and End Results (SEER) 2010–2014. Oncologist 2018, 23, 481–488.
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626.
- Thomas, A.; Rhoads, A.; Pinkerton, E.; Schroeder, M.C.; Conway, K.M.; Hundley, W.G.; McNally, L.R.; Oleson, J.; Lynch, C.F.; Romitti, P.A. Incidence and Survival Among Young Women with Stage I–III Breast Cancer: SEER 2000–2015. JNCI Cancer Spectr. 2019, 3, pkz040.
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692.
- Newman, L.A.; Kaljee, L.M. Health Disparities and Triple-Negative Breast Cancer in African American Women: A Review. JAMA Surg. 2017, 152, 485–493.
- Iqbal, J.; Ginsburg, O.; Rochon, P.A.; Sun, P.; Narod, S.A. Differences in breast cancer stage at diagnosis and cancer-specific survival by race and ethnicity in the United States. JAMA 2015, 313, 165–173.
- Murphy, C.C.; Bartholomew, L.K.; Carpentier, M.Y.; Bluethmann, S.M.; Vernon, S.W. Adherence to adjuvant hormonal therapy among breast cancer survivors in clinical practice: A systematic review. Breast Cancer Res. Treat. 2012, 134, 459–478.
- Puhalla, S.; Bhattacharya, S.; Davidson, N.E. Hormonal therapy in breast cancer: A model disease for the personalization of cancer care. Mol. Oncol. 2012, 6, 222–236.
- Figueroa-Magalhães, M.C.; Jelovac, D.; Connolly, R.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136.
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300.
- Andreopoulou, E.; Schweber, S.J.; Sparano, J.A.; McDaid, H.M. Therapies for triple negative breast cancer. Expert Opin. Pharmacother. 2015, 16, 983–998.
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy with Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909.
- American Cancer Society. Triple-Negative Breast Cancer. Available online: https://www.cancer.org/cancer/types/breast-cancer/about/types-of-breast-cancer/triple-negative.html (accessed on 10 September 2021).
- Berger, E.R.; Park, T.; Saridakis, A.; Golshan, M.; Greenup, R.A.; Ahuja, N. Immunotherapy Treatment for Triple Negative Breast Cancer. Pharmaceuticals 2021, 14, 763.
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098.
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet 1993, 9, 138–141.
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer --Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898.
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123.
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e1021.
- Ciriello, G.; Magnani, L. The many faces of cancer evolution. iScience 2021, 24, 102403.
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer 2016, 2, 49–63.
- O’Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162.
- Wali, V.B.; Langdon, C.G.; Held, M.A.; Platt, J.T.; Patwardhan, G.A.; Safonov, A.; Aktas, B.; Pusztai, L.; Stern, D.F.; Hatzis, C. Systematic Drug Screening Identifies Tractable Targeted Combination Therapies in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 566–578.
- Menden, M.P.; Wang, D.; Mason, M.J.; Szalai, B.; Bulusu, K.C.; Guan, Y.; Yu, T.; Kang, J.; Jeon, M.; Wolfinger, R.; et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 2019, 10, 2674.
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576.
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16.
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e1613.
- Etherington, M.S.; DeMatteo, R.P. Tailored management of primary gastrointestinal stromal tumors. Cancer 2019, 125, 2164–2171.
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714.
- Shen, J.; Li, L.; Howlett, N.G.; Cohen, P.S.; Sun, G. Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells. Cancers 2020, 12, 1087.
- Shen, J.; Li, L.; Yang, T.; Cohen, P.S.; Sun, G. Biphasic Mathematical Model of Cell-Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells. Cancers 2020, 12, 436.
- Di Veroli, G.Y.; Fornari, C.; Goldlust, I.; Mills, G.; Koh, S.B.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. An automated fitting procedure and software for dose-response curves with multiphasic features. Sci. Rep. 2015, 5, 14701.
- Li, L.; Cui, Y.; Shen, J.; Dobson, H.; Sun, G. Evidence for activated Lck protein tyrosine kinase as the driver of proliferation in acute myeloid leukemia cell, CTV-1. Leuk. Res. 2019, 78, 12–20.
- Shen, J.; Li, L.; Yang, T.; Cheng, N.; Sun, G. Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules 2019, 24, 623.
- Ku, G.C.; Chapdelaine, A.G.; Ayrapetov, M.K.; Sun, G. Identification of Lethal Inhibitors and Inhibitor Combinations for Mono-Driver versus Multi-Driver Triple-Negative Breast Cancer Cells. Cancers 2022, 14, 4027.
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55.
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446.
- Medina, P.J.; Goodin, S. Lapatinib: A dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin. Ther. 2008, 30, 1426–1447.
- Tevaarwerk, A.J.; Kolesar, J.M. Lapatinib: A small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin. Ther. 2009, 31 Pt 2, 2332–2348.
- Heerding, D.A.; Rhodes, N.; Leber, J.D.; Clark, T.J.; Keenan, R.M.; Lafrance, L.V.; Li, M.; Safonov, I.G.; Takata, D.T.; Venslavsky, J.W.; et al. Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{oxy}-1H-imidazopyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem. 2008, 51, 5663–5679.
- Levy, D.S.; Kahana, J.A.; Kumar, R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009, 113, 1723–1729.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- O’Neill, S.; Porter, R.K.; McNamee, N.; Martinez, V.G.; O’Driscoll, L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci. Rep. 2019, 9, 3788.
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581.
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046.
- Weiner-Gorzel, K.; Murphy, M. Mitochondrial dynamics, a new therapeutic target for Triple Negative Breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188518.
- Humphries, B.A.; Cutter, A.C.; Buschhaus, J.M.; Chen, Y.C.; Qyli, T.; Palagama, D.S.W.; Eckley, S.; Robison, T.H.; Bevoor, A.; Chiang, B.; et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 60.
- Okada, N.; Ueki, C.; Shimazaki, M.; Tsujimoto, G.; Kohno, S.; Muranaka, H.; Yoshikawa, K.; Takahashi, C. NFYA promotes malignant behavior of triple-negative breast cancer in mice through the regulation of lipid metabolism. Commun. Biol. 2023, 6, 596.
- Pérez-Treviño, P.; Aguayo-Millán, C.D.; Santuario-Facio, S.K.; Vela-Guajardo, J.E.; Salazar, E.; Camacho-Morales, A.; Ortiz, R.; García, N. Metastatic TNBC is closely associated with a fused mitochondrial morphology and a glycolytic and lipogenic metabolism. Biochem. Cell Biol. 2021, 99, 447–456.
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638.
- Davalos, V.; Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J. Clin. 2023, 73, 376–424.
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428.
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159.
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054.
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569.
- Khaled, N.; Bidet, Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers 2019, 11, 559.
- Zolota, V.; Tzelepi, V.; Piperigkou, Z.; Kourea, H.; Papakonstantinou, E.; Argentou, Μ.I.; Karamanos, N.K. Epigenetic Alterations in Triple-Negative Breast Cancer-The Critical Role of Extracellular Matrix. Cancers 2021, 13, 713.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Berdasco, M.; Esteller, M. Towards a druggable epitranscriptome: Compounds that target RNA modifications in cancer. Br. J. Pharmacol. 2022, 179, 2868–2889.
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481.
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873.
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404.
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106.
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021, 20, 789–797.
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
446
Revisions:
2 times
(View History)
Update Date:
09 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No