Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dorota Formanowicz | -- | 4596 | 2023-10-20 11:52:00 | | | |
| 2 | Rita Xu | Meta information modification | 4596 | 2023-10-24 03:40:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gutowski, �.; Kanikowski, S.; Formanowicz, D. Mast Cell Involvement in the Musculoskeletal Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/50612 (accessed on 10 August 2026).
Gutowski �, Kanikowski S, Formanowicz D. Mast Cell Involvement in the Musculoskeletal Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/50612. Accessed August 10, 2026.
Gutowski, Łukasz, Szymon Kanikowski, Dorota Formanowicz. "Mast Cell Involvement in the Musculoskeletal Diseases" Encyclopedia, https://encyclopedia.pub/entry/50612 (accessed August 10, 2026).
Gutowski, �., Kanikowski, S., & Formanowicz, D. (2023, October 20). Mast Cell Involvement in the Musculoskeletal Diseases. In Encyclopedia. https://encyclopedia.pub/entry/50612
Gutowski, Łukasz, et al. "Mast Cell Involvement in the Musculoskeletal Diseases." Encyclopedia. Web. 20 October, 2023.
Copy Citation
There has been a noteworthy revival of interest in the function of mast cells (MCs) in the human body. It is now acknowledged that MCs impact a wide array of processes beyond just allergies, leading to a shift in research direction.
mast cell
MC
rheumatoid arthritis
RA
spondyloarthritis
1. Introduction and Methodology
Since their discovery in the second half of the 19th century, MCs have mainly been studied regarding their role in allergies. However, the turn of the century saw a surge of interest in their impact on diseases related to joints, muscles, and tendons. Nowadays, one can encounter both far-fetched opinions about the crucial role of MCs in the majority of disease entities and opinions trying to tone down this enthusiasm, pointing out many inconsistencies, even errors, in many of the conducted studies.
2. Mast Cells Biology
MCs are far more complex than they are commonly given credit for. Understanding the fundamental functions of these cells is essential to grasp their impact on musculoskeletal conditions. While considered part of the innate immune system, MCs are amongst the most important immune cells sensing danger signals; their role is positioned between the innate and acquired immune systems. While their involvement in immunoglobulin E (IgE)-mediated allergic inflammation is well known, they have also been implicated in various non-allergic inflammatory processes, acting as an early warning system for invaders and coordinating and directing the immune response [1][2]. MCs are developed from CD34+ hematopoietic precursor cells, which occur in the bone marrow and circulate in the blood immaturely. They can fully differentiate and mature only after taking residence in specific tissues. They can be activated by factors such as IgG–antigen complexes, pathogen-associated molecular patterns (PAMPs), complements, cell–cell contact, cytokines, certain drugs, hormones, and physical activators such as temperature and pressure. They reside not only in tissues such as the skin, gut, and respiratory and urinary tract that form the barriers between the self and the environment but also within lymph nodes, near blood vessels, and nerves, giving them the perfect location for early detection and safeguarding the organism [3].
MCs secrete a variety of products: proteases (tryptase, chymase, carboxypeptidase, MCP-1, MCP-2), proteoglycans (heparin, chondroitin sulfate), proteins (CRH, osteopontin, thymic stromal lymphopoietin), biogenic amines (histamine, serotonin), growth factors (PDGF, VEGF, FGF2, bFGF, NGF, PAF), and cytokines (TNFα, lymphotactin IL-1β, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 IL-13, IL-16, IL-17A, IL-18, IL-23 IFN-α, IFN-β, IFN-γ). Moreover, MCs possess the ability to synthesize plasma-membrane-derived lipid mediators, including prostaglandin D2 (PGD2) and leukotriene B4 (LTB4), for various biological actions; in addition, they are proficient antigen-presenting cells [1]. The critical element to release these mediators is degranulation. It is a complex process involving membrane fusion and various proteins, depending on whether it is triggered by allergic or non-allergic reactions [4]. In a non-allergic reaction, degranulation is initiated by the fusion of granules with the plasma membrane, but this process depends on the interaction between vesicle-associated membrane protein 8 (VAMP8) and 7 (VAMP7) exclusively present on the granule membrane of the mast cell. In non-allergic reactions, there are three main pathways of degranulation activation:
- (a)
-
Mas-related G-protein-coupled-ligand receptor member X2 (MRGPRX2) binding results in Inositol Phosphate-Phospholipase C (PI-PLC) activation, thus leading to the formation of Inositol trisphosphate (IP3) and the opening of Ca2+ channels, increasing intracellular Ca2+ concentration and starting degranulation.
- (b)
- (c)
-
Activation by physical contact with activated T-cell membranes and by released microvesicles stimulates extracellular signal-regulated kinase 1/2 (ERK1/2) activation and causes the expression of cytokines, chemokines, adenosine, and growth factors. Adenosine binds to A3 receptors (A3R) and initiates ERK1/2 signaling. It is suspected that neuroblastoma RAS viral oncogene homolog (N-Ras) may be activated by this method, resulting in ERK activation associated with increased cytokine release. Also, this pathway results in tumor necrosis factor-alpha (TNF-α) release, which stimulates the production of the granule-associated enzyme matrix metallopeptidase 9 (MMP-9) [2]. MMP-9 is produced from proMMP-9 with the use of chymase present in mast cells. The stimulation of mast cells by inflammation and oxidative and mechanical stress causes the release of chymase from the granules of mast cells, changing the pH in which chymase is operating from 5,5 in mast cell granules to 7-9, creating ideal conditions for chymase to employ. Moreover, because of endogenous chymase inhibitors (α-antitrypsin and α-antichymotrypsin) found in blood, the enzymatic function of chymase disappears immediately; thus, only activated mast cells can exert its enzymatic function [6].
Additionally, it is pertinent to highlight the interplay between regulatory T cells (T-reg) and mast cells, as their interaction plays a pivotal role in achieving optimal inflammation suppression. This relationship can be described as pseudo-symbiotic, wherein T-reg cells attract mast cells by secreting Interleukin 9 (IL-9), a growth and activation factor for mast cells. In return, mast cells release IL-2, a critical factor for the proliferation and function of T-reg cells. This pathway of mast cell activation was found to be Ig-E-dependent, like in an allergic reaction. A similar connection between T-reg cells, IL-9, and MCs is suspected in immune suppression in B-cell non-Hodgkin’s lymphoma and murine lymphoma. However, the precise mechanism causing such suppression has not been fully discovered yet [2].
In an allergic reaction, mast cells are activated by recognition of an Ag-specific IgE bound to the α subunit of FcεRI expressed on the cell surface. Protein tyrosine kinase Lyn provides a recognition signal allowing for intracellular signal transformation by transphosphorylation of the Fcγ receptor I β (FcεRI β) and γ subunits. Due to alternative splicing, two isoforms of Lyn kinase exist in MCs—Lyn A (56 kDa) and Lyn B (53 kDa). Tyrosine kinases are crucial for MCs in allergic reactions by coimmunoprecipitation with FcεRI, thus leading to the phosphorylation of crucial substances such as Syk tyrosine kinase. This fact allows phospholipase C (PLC)γ to catalyze the hydrolysis of IP3 and, in effect, trigger calcium influx from the extracellular environment via Orai1/CRACM calcium channels. Calcium influx and PLC are essential to release allergic mediators stored in granules, such as histamine and the de novo synthesis of cytokines and eicosanoids by MCs. Lyn B is less effective in triggering calcium responses and MC degranulation but is equivalent to Lyn A in total cellular tyrosine phosphorylation and FcεRI phosphorylation. Lyn A shows more robust calcium responses and degranulation than Lyn B and is better at promoting the interaction of PLCγ with phospho-LAT (see Figure 1) [4][5].
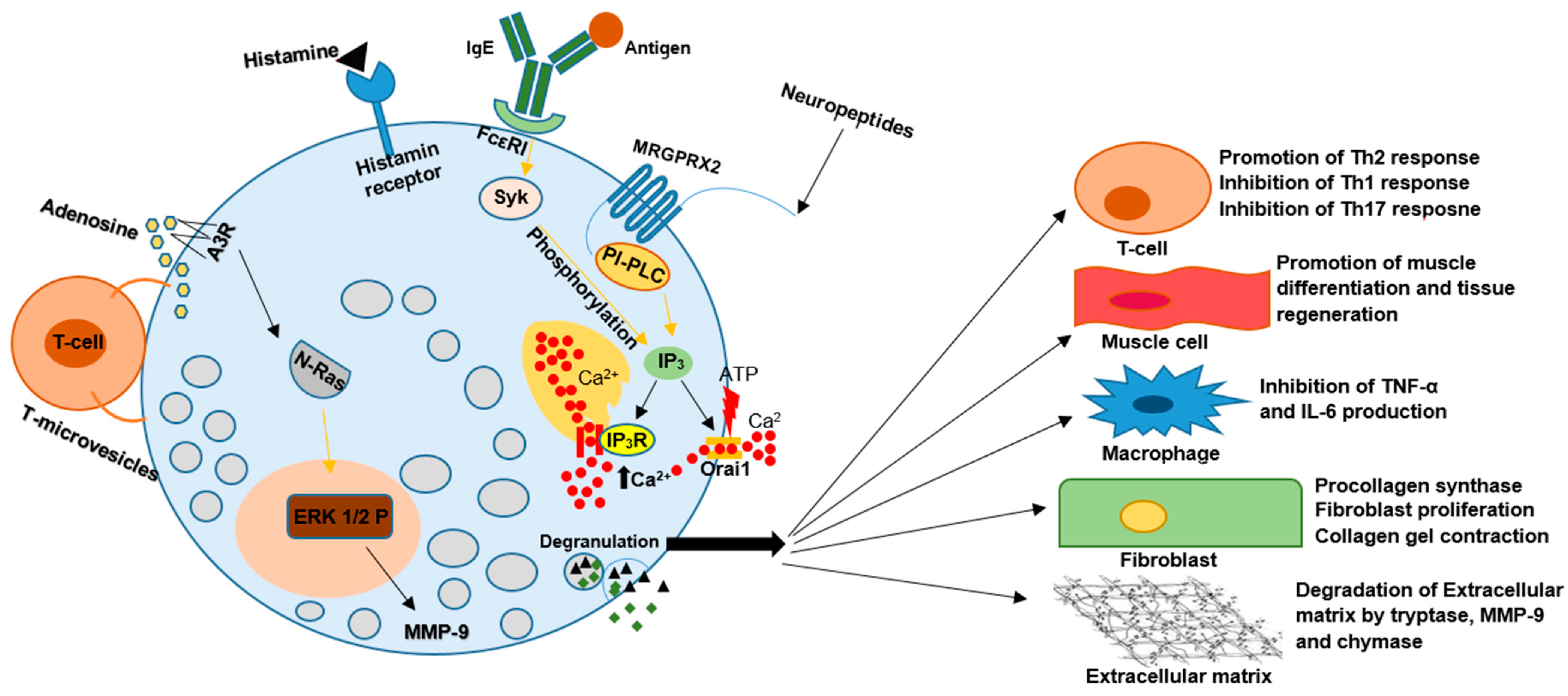
Figure 1. The pleiotropic nature of MCs functions result in their involvement in the pathogenesis of many disease entities. Various factors such as IgG–antigen complexes, PAMPs, complements, physical activators, cell–cell contact, cytokines, certain drugs, neuropeptides, and hormones can activate MCs during degranulation. MCs secrete vasoactive amines, cytokines, and proteases, including tryptase, chymase, and carboxypeptidase. They can also synthesize plasma-membrane-derived lipid mediators, including PGD2 and LTB4. All of the above can change the activity of other immune cells, muscle cells, fibroblasts, and many more. The induced changes have the potential to result in an aggravation or inhibition of the development of many disorders.
The information presented above provides an overview of the fundamental functions of MCs as cells vital in inflammatory and other immune responses. Apart from their well-established association with allergies and increased vascular permeability, MCs also play a role in responding to bacterial and viral infections, parasite infestations, and systemic diseases. Additionally, MCs play a crucial part in tissue remodeling, angiogenesis, and wound healing. There are compelling indications that mast cells are implicated in the pathogenesis of numerous musculoskeletal diseases, such as rheumatoid arthritis, psoriatic arthritis, scleroderma, multiple sclerosis, and many others [1][4]. In inflamed tissues, MCs utilize the aforementioned mediators to modulate the actions of cells like neutrophils or T-cells and present antigens to the latter. MCs can interact with different types of T-cells, thus showing not only proinflammatory but also anti-inflammatory effects. Interestingly, there is even suspected to be a regulatory mast cell population [2]. These dual capabilities of MCs, though not fully understood or well documented, could potentially explain why many high-quality studies report conflicting results regarding the impact of MCs on specific diseases.
3. Rheumatoid Arthritis
Rheumatoid Arthritis (RA) is a chronic autoimmune disease manifested by joint swelling and tenderness, which can progress to disability, impacting both the physical and mental well-being of the patient. It affects 5 to 10 per 1000 people and disproportionally affects the female population [7][8]. Recently, increasing attention has been focused on uncovering the involvement of mast cells in the pathophysiology of this disease. Nevertheless, certain studies conducted on mice have raised some valid concerns.
3.1. Mouse Models
To gain a comprehensive understanding of the challenges associated with animal research on the impact of MCs in RA pathogenesis, it is essential to familiarize oneself with the mouse models commonly employed in many studies and be aware of their limitations. To study RA pathogenesis, MC-deficient mouse strains were generated. The initial MC-deficient mouse type was based on a mutation in the gene encoding receptor tyrosine kinase (Kit). The Kit pathway, initiated by Kit and its ligand SCF, is essential for MC maturation, survival, and activation. Standard models based on this mutation are KitW/Wv, KitW-sh/W-sh, and WCB6F1-MgfSl/Sl-d—all of them result in MC deficiency. Unfortunately, Kit has pleiotropic functions, such as regulating metabolic responses and pain. It also affects the development of hematopoietic stem and progenitor cells, red blood cells, neutrophils, intestinal pacemaker cells, melanocytes, and germ cells. KitW/Wv mice suffer from anemia, neutropenia, and show impairment in lymphocyte development. WCB6F1-MgfSl/Sl-d mice manifest similar abnormalities to the WBB6F1-KitW/Wv mice. KitW-sh/W-sh are fertile and have milder abnormalities, namely splenomegaly, neutrophilia, and thrombocytosis [9]. Kit-independent MC-deficient mice provided researchers with a valuable tool to investigate the precise role of mast cells in immunological and inflammatory reactions. However, it has been proven that the selective absence of mast cells has different consequences than the combined mast cells and Kit deficiency. As Kit exerts a widespread influence on various organism functions, it became challenging to draw definitive conclusions solely on the impact of mast cells on experiments. To distinguish Kit deficiency from MC deficiency, mast-cell-reconstitution Kit mutants have been reconstituted with bone marrow-derived cultured mast cells (BMMCs) [10]. Because of the abovementioned reasons, considerable efforts have been dedicated to generating mouse strains that are exclusively deficient in mast cells without affecting other immune system components.
The Cre-mediated MC eradication mouse line (Cre-Master, Cpa3Cre/+) was generated by targeted insertion of Cre-recombinase into the mast cell Cpa3 gene. High expression of the Cre recombinase exerts genotoxicity, leading to MC eradication. Except for the reduction in basophil level, mice still have a proper immune system but cannot initiate IgE-mediated allergic responses. Nevertheless, they are fully vulnerable to antibody-induced autoimmune arthritis. Similarly, mouse Cpa3Cre; Mcl-1fl/fl strain (Cpa3-Cre) has a strongly reduced number of MCs (92% to 100%), a milder decrease in the number of basophils (58% to 78%), but impaired IgE-mediated responses [9][10].
To develop a strain with no abnormalities in the cellular composition of the spleen, blood, skin, and bone marrow, the Mcpt5Cre mouse line was mated with the R-DTA strain. Mcpt5Cre R-DTA mice resulted in the MC-specific expression of DTA and, thus, in a toxin-mediated constitutive reduction of 90% of MCs in the peritoneum and skin [9].
An opposite approach was used to generate mice lacking A20—specifically, in connective-tissue-type MCs (Mcpt5Cre A20F/F). The MC-specific ablation of A20, a negative feedback regulator, exaggerates inflammatory MC signaling but does not provoke anaphylactic reactions. As it turns out, hyperactive A20-deficient MCs cause an earlier onset and worsening of Collagen-Induced Arthritis (CIA) symptoms [11].
To date, numerous methods for inducing arthritis to simulate RA have been developed and investigated in the aforementioned mouse models. One of them is the transfer of serum from K/BxN transgenic mice (serum-transfer-induced arthritis, STIA), which produce autoantibodies against the glucose-6-phosphate isomerase (GP6I), inducing an arthritic phenotype. KitW/Wv and MgfSl/Sl-d mice are protected from arthritis induced in such a way, but reconstituting KitW/Wv mice with BMMC brought back the susceptibility to the disease. It was also reported that neutrophil transfer could be sufficient to restore K/BxN-passive arthritis in KitW/Wv mice. Some of the disagreements could be a consequence of how many arthritogenic antibodies were given to each animal and on what day the arthritis was evaluated. Despite that, KitW-sh/W-sh mice were not protected from STIA, probably because of impaired neutrophil levels. Similar conclusions were drawn by Pitman et al.; in their research, collagen-induced arthritis (CIA), which is induced by immunizing with type II collagen, was used in wild-type and MC-deficient KitW-sh/W-sh mice on a C57BL/6 background. The absence of mast cells did not substantially change the disease course. Therefore, immune activation against type II collagen can drive mast-cell-independent pathways, leading to synovitis in KitW-sh/W-sh mice [12][13][14][15].
Schubert et al. identified problems within the studies carried out on Cre-mediated mice. According to their research, MC-competent Cre−; iDTR+ mice and MC-depleted cpt5-Cre+; iDTR+ mice, which received K/BxN sera, showed equal levels of arthritis. However, in STIA, arthritis is induced in a T-cell-independent manner, while in humans, RA can be activated in both humoral and cellular ways. Nonetheless, the same mice after the induction of CIA proved MCs to be critically relevant, not only because the absence of MCs resulted in lower inflammation but also in the loss of T cell expansion and reduced T cell cytokine levels [16]. However, Cunin et al. stated that the mechanisms behind mouse models of MC influence are much more nuanced, and the role of MCs in a particular setting can vary depending on the need for amplifying the underlying inflammatory trigger. It is also possible to learn from the same work about another critical factor—megakaryocytes, which, through IL-1 secretion, can activate synovial fibroblasts. The presence of these factors restores the susceptibility of KitW/Wv mice to RA induction [17].
3.2. MCs Types in RA
MCs are resident cells in the healthy synovial joint, but in the synovium of RA patients, MC hyperplasia can be observed [18], although this feature is common in many autoimmune diseases [19]. Recent studies examining the synovium in early RA have revealed distinct synovitis phenotypes based on MC infiltration density [18]. Patients with high MC presence have substantially increased CRP levels, high lymphocyte counts in the RA synovium, and high disease activity scores. However, data concerning the correlation of ESR with the amount of MC are contradictory and study-dependent [18][20][21]. Additionally, two genes regarded as RA’s diagnostic markers—LSP1 and GNLY—are also associated with MCs, underscoring the relevance of these cells in RA pathogenesis [8].
Nevertheless, the detailed impact on the RA mechanism has not been fully elucidated; even their role in the different phases of RA development remains a topic of debate [22]. Two main types of mast cells can be distinguished: the tryptase-chymase double-positive cell (MCTC) and tryptase-only positive cell (MCT). MCT is associated with inflammation, especially in the early stages of RA, while MCT expansion followed by MCTCs is associated with tissue remodeling, characteristic of established or chronic disease. MCTCs are also the primary subtype in the normal synovium [19]. It noteworthy that RA patients in ultrasound-defined remission had significantly reduced synovial mast cell density compared to patients with clinically active RA [23].
3.3. Autoantibodies and Receptors
In rheumatoid arthritis, anti-citrullinated protein antibodies (ACPA) are an essential group of autoantibodies. They recognize various peptides in which arginine has been modified into citrulline by the family of Peptidyl Arginine Deiminase (PAD) enzymes, which can be produced in RA by macrophages, dendritic cells, neutrophils, and mast cells [24].
ACPA can bind to antigens and activate the complement system, activating mast cells through the cleavage product C5a. It is unclear if this pathway leads to autoantibody-mediated mast cell activation in humans. Mast cells can also be activated directly by Fc receptors, particularly Fcγ receptors. Because ACPAs are mostly IgM and IgG isotypes, IgG-ACPA binding to receptors is thought to play a significant role in autoantibody-induced pathogenesis [19]. IL-33 also enhances this activation by inducing MCs to release IL-10, histamine, and other cytokines associated with type 2 immune responses, such as IL-5 and IL-13 [25].
Mice synovial MCs express the activating FcγRIIIa, which is involved in arthritis induced by the anti-collagen autoantibodies model [19]. In contrast, the Fc receptor β chain, also an FcγRIII component, negatively regulates arthritic inflammation in a mice knock-out model [26]. Nevertheless, human studies showed that synovial MCs of all studied patients could be activated by the cross-linking of ACPA with another receptor—FcγRIIA—which suggests that this is a significant factor in autoantibody-mediated mast cell activation [19]. It agrees with the study by Lee et al., according to which FcRI and FcRII are responsible for the induction of aggregated IgG-dependent activation of synovial MCs obtained from patients with RA [27] and with the study conducted by Suurmond et al., where both human-cultured and synovial mast cells expressed FcγRIIA. However, in the latter, FcγRI was not expressed by synovial mast cells derived from most patients [28].
Nonetheless, it should be taken into consideration that Tsuboi et al. reported that in human-FcγRIIa-expressing, γ-chain-deficient (hFcγRIIa+/γ−/−) mice, stimulation of neutrophils via FcRIIa is not associated with mast cell degranulation and, indeed, proceeds in the absence of these cells [27]. Also, according to Mancardi et al., at least IgG1 and IgG2 antibody isotypes, and two receptors, FcgRIIIA and FcgRIV, contribute to K/BxN-induced arthritis, and at least two cell types other than mast cells are required for this process—monocytes or macrophages, and neutrophils [15].
The molecular processes taking place between FCγ activation and MC degranulation are insufficiently studied. Guma et al. demonstrated using K/BxN C-Jun N-terminal kinase (JNK) 1−/− and Jnk2−/− mice) that JNK1 but not JNK2 is an essential component of FcγR-induced release of proinflammatory mediators, such as IL-1β [29].
Other receptors involved in RA are Toll-like receptors (TLRs). Mast cells express a variety of TLRs, which induce MC activation after being triggered. MCs also express TLR-mediated responses to endogenous ligands released during inflammation, such as TLR-2, TLR-4, and endosomal TLRs [19].
TLR4-mediated signals induce the production of IL-12 by macrophages, mast cells, and Gr-1+ cells, which regulate IL-1b and IFN-g production, suppressing TGF-β production. This regulation of the cytokine network plays a crucial role in joint inflammation [30].
The combination of TLR- and Fcγ-mediated activation enhances cytokine production by human mast cells. Such synergy can dictate the type and extent of immune cells attracted to the inflammation site. The synergy between TLRs or cytokines and the Fc receptor is highly beneficial when pathogen elimination is required but can also lead to the further release of modified autoantigens and TLR ligands, contributing to the chronicity of rheumatoid arthritis [19][28].
3.4. Cytokines
Cytokines can not only affect mast cells’ survival but can also activate them. Cytokines such as IL-3, IL-4, IL-5, and IL-33, which are increased in synovial fluid of RA patients, can influence MC proliferation and activity. The stimulation of MCs with cytokines alone contributes more to their proliferation than activation, but the activation effect combined with other factors is powerful. Recently, great attention has been paid to IL-33, which has been shown to have multiple implications on MC functionality. IL-33 is derived mainly from synovial fibroblasts and can regulate the maturation and activation of MC on its own, increasing the number of tryptase-positive and tryptase-chymase double-positive cells [31][32][33]. Via its receptor ST2, lL-33 induces the expression of the activating receptor for the Fc fragment of IgG (FcgRIIa) in human mast cells [25]. It is worth noticing that in vivo, IL-33 exacerbated CIA in ST2−/− mice engrafted with WT but not ST2−/− mast cells [34]. Furthermore, IL-33 also induces osteoclast differentiation from synovial fluid monocytes independent of RANKL but reduces their differentiation in RANKL-induced osteoclastogenesis. IL-33-stimulated MCs induce more osteoclast differentiation, probably by cell–cell contact [32]. Nonetheless, some studies indicate that IL-33 mRNA levels were inversely correlated with proinflammatory markers, such as CD14, CD16 (FcgRIIIA), and TNF. These observations speculate that mast cells, particularly when triggered by IL-33, could dampen the activation of monocytes and modulate the immune response in the synovial tissue of patients with RA [25][35].
Another essential cytokine is IL17A (also called IL-17), which is elevated in the synovial fluid of RA patients. It has been considered lately as one of the potential new targets for treating this disease. IL-17 can induce the production of proinflammatory factors such as IL-6, IL-1, TNF, and matrix metalloproteinases. While, in the peripheral blood of patients with immune-mediated disease, Th17 cells are the most critical source of IL-17, in the joints of RA patients, mast cells have been considered essential producers of this interleukin [36]. This thesis was also confirmed by Hueber et al., who proved that MCs are one of the primary sources of IL17A in established synovitis [37], but in vitro studies did not show such a relationship [38].
3.5. Granule Proteins
MCs secrete many products, but some of them—like chymase, histamine, and tryptase—are elevated in both the synovial fluid and peripheral blood of RA patients. Since they are mast-cell-specific, it likely reflects local mast cell activation, which is supported by the fact that serum tryptase is correlated with RA disease activity [19][32]. It is believed that many serine proteinases are thought to be involved in rheumatoid arthritis as they are elevated in the synovial fluid of such patients. Proteinases transmit the signal through the Protease-Activated Receptors (PAR) receptors family, allowing them to function as inflammatory mediators and degradative enzymes. Depending on the PAR family member, PAR activation can modulate inflammation and pain. As it turns out, in rats, PAR4 is expressed on synovial MCs, which indicates that they are key players in mediating the signaling effects of serine proteinases. In this case, PAR4 activation resulted in a pronociceptive effect depending on mast cell activation [39].
Tryptase is one of the serine proteinases, characteristic for MC, and it is the main substance stored in MCT (MCs containing only tryptase). Tryptase, along with histamine, is co-responsible for increased vascular permeability during inflammation, and fibroblast and epithelial cell proliferation. It is also one of the compounds involved in MC–neuron communication. As it turns out, injection of this protease into the mice joints results in inflammation and swelling; corresponding studies have proven that mice lacking monocyte chemotactic protein 6, a human tryptase analog, show resistance to antibody-mediated arthritis. That is why tryptase and its receptor, PAR-2, may play a significant role in RA pathogenesis.
This information sheds light on two processes co-occurring in the tissues of RA patients: the proliferation of RA synovial fibroblasts (RASFs), which contributes to the pathogenesis of RA, and increased production of Fas, which should lead to apoptosis of RASFs. Although in vitro Fas-mediated apoptosis is significant, in vivo, this mechanism of apoptosis induction is limited. This seemingly contradictory information was reconciled when it turned out that RASFs express the receptor for mast cell tryptase (PAR-2) and, thus, are protected from Fas-mediated apoptosis by tryptase in a Rho-kinase-dependent manner. PAR-2 is also expressed in other cells; therefore, it should be investigated if MCs affect them through a tryptase/PAR-2-dependent mechanism in RA patients [4][40].
In addition to tryptase, MCs secrete other serine proteinases, such as chymase and carboxypeptidase A3. Chymase can degrade the extracellular matrix, activate procollagenases, aggravate leukocyte migration, and affect tissue remodeling in diseases like asthma and chronic obstructive pulmonary disease [4]. MC-chymase does not significantly affect normal synovial fibroblast (SFB) growth but induces the proliferation of fibroblast-derived from rats with CIA. CIA-SFB treated with chymase exhibits increased MMP-9 and lowered MMP-2 expression. Because of altered MMP-9 levels, chymase promotes CIA-SFB adherent abilities and invasive migration [41]. Similar conclusions regarding MMP-9 can be drawn based on mouse models in which mMCP-5, an enzyme similar to human chymase, also affected levels of this metalloproteinase [42]. Carboxypeptidase A3 (CPA3), also known as human MC carboxypeptidase A, is a protease highly expressed in MCs. CPA3 is stored in their granules by ionically bounding to heparin-containing serglycin proteoglycans. Besides its primary proteolytic function, CPA3, like the other mediators in MC granules, also affects inflammation, pain, vasodilation, angiogenesis, coagulation, tissue remodeling, and pro-hormone/enzyme cleavage. Notably, CPA3 is a characteristic feature of MCtc (MC type containing tryptase, chymase, and CPA3), which allows it to distinguish this type of MC from MCt [4][13][42].
Another vital substance stored in mast cell granules is histamine. It is an essential mediator, acting via four receptors (HR1-4). All of them have vastly different effects. HR-2 is a receptor antagonizing functions of HR1. It also reduces histamine release in basophils and MCs. However, studies on respective gene-deficient mice show that HR1- and HR2-deficient mice are unprotected from auto-antibody-induced arthritis. HR3 regulates the release of histamine and the negative feedback mechanism responsible for reducing central histaminergic activity. There is seemingly an HR-3-controlled mast-cell–neuron feedback loop. HR-4, a receptor very similar to HR-3, is involved in the recruitment and activation of inflammatory cells like mast cells, neutrophils, eosinophils, dendritic cells, and T cells. Thus, it is perceived as the immune system histamine receptor. Interestingly, HR-4 attracts mast cells toward histamine without causing their degranulation. In the K/B×N-antibody-induced arthritis model administration of the HR4 antagonist, clozapine protects mice from arthritic symptoms, underscoring its critical role as the primary histamine receptor in arthritis. On the other hand, inhibiting HR-3 has a slight initial effect on the mentioned disorder. However, it is important to note that the HR-3 antagonist, thioperamide, partially blocks HR-4, as it shares about 40% homology with HR3 [43][44].
3.6. Dependencies between MCs and Other Immune Cells in RA
In recent years, it has been proven that MCs can affect T lymphocytes in several ways. One of the well-known functionalities of MCs is the ability to present antigens to CD4+ T-cells. Mast-cell-derived cytokines can also induce T-cell activation. It has also been proven that the interaction between T helper cells and mast cells not only activates T-cells but can also change MC phenotype; moreover, regulatory T-cells can even inhibit MC activation [19].
In vitro studies suggest that activated MCs can also increase B cell activation, proliferation, and differentiation into IgM- and IgG-producing B cells, as well as enhance their antigen-presenting capacity. There is no certainty about the exact mechanism by which MCs induce B cells, but one of the confirmed means is cell contact and, specifically, CD40L–CD40 interaction [20][45]. Another proposed means of B cell activation by MC is Bruton’s tyrosine kinase (BTK), which acts as a proinflammatory in the synovium of RA patients. Activated BTK signaling can also increase the expression of proinflammatory cytokines, chemokines, and cell adhesion molecules [46][47].
In RA synovial tissue, human synovial MCs can be found close to CD141 monocyte/macrophages, CD31 T cells, and CD201 B cells. They were most commonly spotted near macrophages. MCs activated with IL-33 and IgG through the release of IL-10 and histamine were able to suppress monocyte activation by inhibiting both TNF production and up-regulation of the costimulatory molecule CD80 [25].
The impact of mast cells on inflammation, achieved through increased vascular permeability and the secretion of chemokines, which in turn leads to the infiltration of neutrophils and other immune cells, is well established. In rheumatoid arthritis, neutrophil chemoattraction to the synovial fluid is mainly mediated by IL-8, which is primarily secreted by mast cells in response to ACPA autoantibodies and TLR ligands. It is suspected that mast cells are helpful not only in enhancing lymphocytic infiltration but also in supporting the maintenance of specific lymphocyte subtypes by secreting growth factors like GM-CSF and G-CSF [19]. Interestingly, studies conducted on DBA/1J mice after immunization with bovine type II collagen suggest that the aforementioned neutrophil influx might vary with time across different joints [48].
References
- Sigal, L.H. Basic Science for the Clinician 53: Mast Cells. JCR J. Clin. Rheumatol. 2011, 17, 395–400.
- Mekori, Y.A.; Hershko, A.Y. T Cell-Mediated Modulation of Mast Cell Function: Heterotypic Adhesion-Induced Stimulatory or Inhibitory Effects. Front. Immunol. 2012, 3, 6.
- Walker, M.E.; Hatfield, J.K.; Brown, M.A. New Insights into the Role of Mast Cells in Autoimmunity: Evidence for a Common Mechanism of Action? Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2012, 1822, 57–65.
- Singh, J.; Shah, R.; Singh, D. Targeting Mast Cells: Uncovering Prolific Therapeutic Role in Myriad Diseases. Int. Immunopharmacol. 2016, 40, 362–384.
- Alvarez-Errico, D.; Yamashita, Y.; Suzuki, R.; Odom, S.; Furumoto, Y.; Yamashita, T.; Rivera, J. Functional Analysis of Lyn Kinase A and B Isoforms Reveals Redundant and Distinct Roles in FcεRI-Dependent Mast Cell Activation. J. Immunol. 2010, 184, 5000–5008.
- Takai, S.; Jin, D. Pathophysiological Role of Chymase-Activated Matrix Metalloproteinase-9. Biomedicines 2022, 10, 2499.
- Zhao, J.; Guo, S.; Schrodi, S.J.; He, D. Molecular and Cellular Heterogeneity in Rheumatoid Arthritis: Mechanisms and Clinical Implications. Front. Immunol. 2021, 12, 790122.
- Yu, R.; Zhang, J.; Zhuo, Y.; Hong, X.; Ye, J.; Tang, S.; Zhang, Y. Identification of Diagnostic Signatures and Immune Cell Infiltration Characteristics in Rheumatoid Arthritis by Integrating Bioinformatic Analysis and Machine-Learning Strategies. Front. Immunol. 2021, 12, 724934.
- Yu, X.; Kasprick, A.; Petersen, F. Revisiting the Role of Mast Cells in Autoimmunity. Autoimmun. Rev. 2015, 14, 751–759.
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Möller, P.; Benoist, C.; Mathis, D.; et al. Cre-Mediated Cell Ablation Contests Mast Cell Contribution in Models of Antibody- and T Cell-Mediated Autoimmunity. Immunity 2011, 35, 832–844.
- Heger, K.; Fierens, K.; Vahl, J.C.; Aszodi, A.; Peschke, K.; Schenten, D.; Hammad, H.; Beyaert, R.; Saur, D.; Van Loo, G.; et al. A20-Deficient Mast Cells Exacerbate Inflammatory Responses In Vivo. PLoS Biol. 2014, 12, e1001762.
- Pitman, N.; Asquith, D.L.; Murphy, G.; Liew, F.Y.; McInnes, I.B. Collagen-Induced Arthritis Is Not Impaired in Mast Cell-Deficient Mice. Ann. Rheum. Dis. 2011, 70, 1170–1171.
- Adachi, R.; Krilis, S.A.; Nigrovic, P.A.; Hamilton, M.J.; Chung, K.; Thakurdas, S.M.; Boyce, J.A.; Anderson, P.; Stevens, R.L. Ras Guanine Nucleotide-Releasing Protein-4 (RasGRP4) Involvement in Experimental Arthritis and Colitis. J. Biol. Chem. 2012, 287, 20047–20055.
- Elliott, E.R.; Van Ziffle, J.A.; Scapini, P.; Sullivan, B.M.; Locksley, R.M.; Lowell, C.A. Deletion of Syk in Neutrophils Prevents Immune Complex Arthritis. J. Immunol. 2011, 187, 4319–4330.
- Mancardi, D.A.; Jönsson, F.; Iannascoli, B.; Khun, H.; Van Rooijen, N.; Huerre, M.; Daëron, M.; Bruhns, P. Cutting Edge: The Murine High-Affinity IgG Receptor FcγRIV Is Sufficient for Autoantibody-Induced Arthritis. J. Immunol. 2011, 186, 1899–1903.
- Schubert, N.; Dudeck, J.; Liu, P.; Karutz, A.; Speier, S.; Maurer, M.; Tuckermann, J.; Dudeck, A. Mast Cell Promotion of T Cell-Driven Antigen-Induced Arthritis Despite Being Dispensable for Antibody-Induced Arthritis in Which T Cells Are Bypassed: Mast Cell Promotion of Antigen-Induced Arthritis. Arthritis Rheumatol. 2015, 67, 903–913.
- Cunin, P.; Penke, L.R.; Thon, J.N.; Monach, P.A.; Jones, T.; Chang, M.H.; Chen, M.M.; Melki, I.; Lacroix, S.; Iwakura, Y.; et al. Megakaryocytes Compensate for Kit Insufficiency in Murine Arthritis. J. Clin. Investig. 2017, 127, 1714–1724.
- Min, H.K.; Kim, K.-W.; Lee, S.-H.; Kim, H.-R. Roles of Mast Cells in Rheumatoid Arthritis. Korean J. Intern. Med. 2020, 35, 12–24.
- Suurmond, J.; Van Der Velden, D.; Kuiper, J.; Bot, I.; Toes, R.E.M. Mast Cells in Rheumatic Disease. Eur. J. Pharmacol. 2016, 778, 116–124.
- Rivellese, F.; Mauro, D.; Nerviani, A.; Pagani, S.; Fossati-Jimack, L.; Messemaker, T.; Kurreeman, F.A.S.; Toes, R.E.M.; Ramming, A.; Rauber, S.; et al. Mast Cells in Early Rheumatoid Arthritis Associate with Disease Severity and Support B Cell Autoantibody Production. Ann. Rheum. Dis. 2018, 77, 1773–1781.
- Rivellese, F.; Rossi, F.W.; Giorli, G.; Napolitano, F.; De Paulis, A.; Pitzalis, C. Persistence of Mast Cell-Positive Synovitis in Early Rheumatoid Arthritis Following Treatment with Conventional Synthetic Disease Modifying Anti-Rheumatic Drugs. Front. Pharmacol. 2020, 11, 1051.
- Van Der Velden, D.; Lagraauw, H.M.; Wezel, A.; Launay, P.; Kuiper, J.; Huizinga, T.W.J.; Toes, R.E.M.; Bot, I.; Stoop, J.N. Mast Cell Depletion in the Preclinical Phase of Collagen-Induced Arthritis Reduces Clinical Outcome by Lowering the Inflammatory Cytokine Profile. Arthritis Res. Ther. 2016, 18, 138.
- Ramírez, J.; Celis, R.; Usategui, A.; Ruiz-Esquide, V.; Faré, R.; Cuervo, A.; Sanmartí, R.; Pablos, J.L.; Cañete, J.D. Immunopathologic Characterization of Ultrasound-Defined Synovitis in Rheumatoid Arthritis Patients in Clinical Remission. Arthritis Res. Ther. 2016, 18, 74.
- Arandjelovic, S.; McKenney, K.R.; Leming, S.S.; Mowen, K.A. ATP Induces Protein Arginine Deiminase 2-Dependent Citrullination in Mast Cells through the P2X7 Purinergic Receptor. J. Immunol. 2012, 189, 4112–4122.
- Rivellese, F.; Suurmond, J.; Habets, K.; Dorjée, A.L.; Ramamoorthi, N.; Townsend, M.J.; De Paulis, A.; Marone, G.; Huizinga, T.W.J.; Pitzalis, C.; et al. Ability of Interleukin-33- and Immune Complex-Triggered Activation of Human Mast Cells to Down-Regulate Monocyte-Mediated Immune Responses: Mast Cell Suppression of Monocyte Activation. Arthritis Rheumatol. 2015, 67, 2343–2353.
- Ohtsubo-Yoshioka, M.; Nunomura, S.; Kataoka, T.R.; Okayama, Y.; Ra, C. Fc Receptor Beta Chain Deficiency Exacerbates Murine Arthritis in the Anti-Type II Collagen Antibody-Induced Experimental Model. Mod. Rheumatol. 2013, 23, 804–810.
- Lee, H.; Kashiwakura, J.-I.; Matsuda, A.; Watanabe, Y.; Sakamoto-Sasaki, T.; Matsumoto, K.; Hashimoto, N.; Saito, S.; Ohmori, K.; Nagaoka, M.; et al. Activation of Human Synovial Mast Cells from Rheumatoid Arthritis or Osteoarthritis Patients in Response to Aggregated IgG through Fcγ Receptor I and Fcγ Receptor II. Arthritis Rheum. 2013, 65, 109–119.
- Suurmond, J.; Rivellese, F.; Dorjée, A.L.; Bakker, A.M.; Rombouts, Y.J.P.C.; Rispens, T.; Wolbink, G.; Zaldumbide, A.; Hoeben, R.C.; Huizinga, T.W.J.; et al. Toll-like Receptor Triggering Augments Activation of Human Mast Cells by Anti-Citrullinated Protein Antibodies. Ann. Rheum. Dis. 2015, 74, 1915–1923.
- Guma, M.; Kashiwakura, J.; Crain, B.; Kawakami, Y.; Beutler, B.; Firestein, G.S.; Kawakami, T.; Karin, M.; Corr, M. JNK1 Controls Mast Cell Degranulation and IL-1β Production in Inflammatory Arthritis. Proc. Natl. Acad. Sci. USA 2010, 107, 22122–22127.
- Kim, H.; Chung, D. TLR4-Mediated IL-12 Production Enhances IFN-γ and IL-1β Production, Which Inhibits TGF-β Production and Promotes Antibody-Induced Joint Inflammation. Arthritis Res. Ther. 2012, 14, R210.
- Xu, D.; Jiang, H.-R.; Li, Y.; Pushparaj, P.N.; Kurowska-Stolarska, M.; Leung, B.P.; Mu, R.; Tay, H.K.; McKenzie, A.N.J.; McInnes, I.B.; et al. IL-33 Exacerbates Autoantibody-Induced Arthritis. J. Immunol. 2010, 184, 2620–2626.
- Kim, K.-W.; Kim, B.-M.; Won, J.-Y.; Min, H.-K.; Lee, K.-A.; Lee, S.-H.; Kim, H.-R. Regulation of Osteoclastogenesis by Mast Cell in Rheumatoid Arthritis. Arthritis Res. Ther. 2021, 23, 124.
- Nile, C.J.; Barksby, E.; Jitprasertwong, P.; Preshaw, P.M.; Taylor, J.J. Expression and Regulation of Interleukin-33 in Human Monocytes: IL-33 in Monocytes. Immunology 2010, 130, 172–180.
- Liew, F.Y. IL-33: A Janus Cytokine: Table 1. Ann. Rheum. Dis. 2012, 71, i101–i104.
- Shakerian, L.; Kolahdooz, H.; Garousi, M.; Keyvani, V.; Kamal Kheder, R.; Abdulsattar Faraj, T.; Yazdanpanah, E.; Esmaeili, S.-A. IL-33/ST2 Axis in Autoimmune Disease. Cytokine 2022, 158, 156015.
- Kenna, T.J.; Brown, M.A. The Role of IL-17-Secreting Mast Cells in Inflammatory Joint Disease. Nat. Rev. Rheumatol. 2013, 9, 375–379.
- Hueber, A.J.; Asquith, D.L.; Miller, A.M.; Reilly, J.; Kerr, S.; Leipe, J.; Melendez, A.J.; McInnes, I.B. Cutting Edge: Mast Cells Express IL-17A in Rheumatoid Arthritis Synovium. J. Immunol. 2010, 184, 3336–3340.
- Kan, J.; Mishima, S.; Kashiwakura, J.; Sasaki-Sakamoto, T.; Seki, M.; Saito, S.; Ra, C.; Tokuhashi, Y.; Okayama, Y. Interleukin-17A Expression in Human Synovial Mast Cells in Rheumatoid Arthritis and Osteoarthritis. Allergol. Int. 2016, 65, S11–S16.
- Russell, F.A.; Zhan, S.; Dumas, A.; Lagarde, S.; Pouliot, M.; McDougall, J.J. The Pronociceptive Effect of Proteinase-Activated Receptor-4 Stimulation in Rat Knee Joints Is Dependent on Mast Cell Activation. Pain 2011, 152, 354–360.
- Sawamukai, N.; Yukawa, S.; Saito, K.; Nakayamada, S.; Kambayashi, T.; Tanaka, Y. Mast Cell-Derived Tryptase Inhibits Apoptosis of Human Rheumatoid Synovial Fibroblasts via Rho-Mediated Signaling. Arthritis Rheum. 2010, 62, 952–959.
- Chu, Y.; Wang, J.; Zhou, X. Mast Cell Chymase in Synovial Fluid of Collagen-Induced-Arthritis Rats Regulates Gelatinase Release and Promotes Synovial Fibroblasts Proliferation via FAK/P21 Signaling Pathway. Biochem. Biophys. Res. Commun. 2019, 514, 336–343.
- Stevens, R.L.; McNeil, H.P.; Wensing, L.A.; Shin, K.; Wong, G.W.; Hansbro, P.M.; Krilis, S.A. Experimental Arthritis Is Dependent on Mouse Mast Cell Protease-5. J. Biol. Chem. 2017, 292, 5392–5404.
- Mehta, P.; Miszta, P.; Rzodkiewicz, P.; Michalak, O.; Krzeczyński, P.; Filipek, S. Enigmatic Histamine Receptor H4 for Potential Treatment of Multiple Inflammatory, Autoimmune, and Related Diseases. Life 2020, 10, 50.
- Nent, E.; Frommholz, D.; Gajda, M.; Bräuer, R.; Illges, H. Histamine 4 Receptor Plays an Important Role in Auto-Antibody-Induced Arthritis. Int. Immunol. 2013, 25, 437–443.
- Palm, A.-K.E.; Garcia-Faroldi, G.; Lundberg, M.; Pejler, G.; Kleinau, S. Activated Mast Cells Promote Differentiation of B Cells into Effector Cells. Sci. Rep. 2016, 6, 20531.
- Hartkamp, L.M.; Fine, J.S.; Van Es, I.E.; Tang, M.W.; Smith, M.; Woods, J.; Narula, S.; DeMartino, J.; Tak, P.P.; Reedquist, K.A. Btk Inhibition Suppresses Agonist-Induced Human Macrophage Activation and Inflammatory Gene Expression in RA Synovial Tissue Explants. Ann. Rheum. Dis. 2015, 74, 1603–1611.
- Watterson, S.H.; De Lucca, G.V.; Shi, Q.; Langevine, C.M.; Liu, Q.; Batt, D.G.; Beaudoin Bertrand, M.; Gong, H.; Dai, J.; Yip, S.; et al. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-Fluoro-1-Methyl-2,4-Dioxo-1,2-Dihydroquinazolin-3(4 H)-Yl)-2-Methylphenyl)-2-(S)-(2-Hydroxypropan-2-Yl)-2,3,4,9-Tetrahydro-1 H -Carbazole-8-Carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J. Med. Chem. 2016, 59, 9173–9200.
- Pimentel, T.A.; Sampaio, A.L.F.; D’Acquisto, F.; Perretti, M.; Oliani, S.M. An Essential Role for Mast Cells as Modulators of Neutrophils Influx in Collagen-Induced Arthritis in the Mouse. Lab. Investig. 2011, 91, 33–42.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
533
Revisions:
2 times
(View History)
Update Date:
24 Oct 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No