Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Magdalena Kuras | -- | 3536 | 2023-10-05 11:28:49 | | | |
| 2 | Lindsay Dong | Meta information modification | 3536 | 2023-10-06 15:00:17 | | | | |
| 3 | Lindsay Dong | -1 word(s) | 3535 | 2023-10-06 15:01:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kuras, M. The Interaction between Melanoma Cells and Tumor Microenvironment. Encyclopedia. Available online: https://encyclopedia.pub/entry/49840 (accessed on 24 June 2026).
Kuras M. The Interaction between Melanoma Cells and Tumor Microenvironment. Encyclopedia. Available at: https://encyclopedia.pub/entry/49840. Accessed June 24, 2026.
Kuras, Magdalena. "The Interaction between Melanoma Cells and Tumor Microenvironment" Encyclopedia, https://encyclopedia.pub/entry/49840 (accessed June 24, 2026).
Kuras, M. (2023, October 05). The Interaction between Melanoma Cells and Tumor Microenvironment. In Encyclopedia. https://encyclopedia.pub/entry/49840
Kuras, Magdalena. "The Interaction between Melanoma Cells and Tumor Microenvironment." Encyclopedia. Web. 05 October, 2023.
Copy Citation
Malignant melanoma is a very aggressive skin cancer, characterized by a heterogeneous nature and high metastatic potential. The incidence of melanoma is continuously increasing worldwide, and it is one of the most common cancers in young adults. The understanding of melanoma biology has increased profoundly, and disease management for patients with disseminated disease has improved due to the emergence of immunotherapy and targeted therapy. However, a significant fraction of patients relapse or do not respond adequately to treatment. This can partly be explained by the complex signaling between the tumor and its microenvironment, giving rise to melanoma phenotypes with different patterns of disease progression.
malignant melanoma
tumor microenvironment
treatment resistance
1. Introduction
Melanoma, a word derived from the Greek melas “dark” and oma “tumor”, emerges from melanocytes, a type of specialized dendritic cells of neural crest origin. Melanocytes can be found in the epidermis of the skin, along the choroidal layer of the eye, on mucosal surfaces, and in the meninges [1]. Cutaneous melanoma has a heterogeneous nature and a strong propensity to metastasize to other organs [2][3]. The continuous increase in melanoma prevalence is becoming a major clinical problem and will be associated with even higher treatment costs in the coming years. According to WHO (2020) and GLOBOCAN (2020), melanoma incidence and mortality due to melanoma are the highest in Australia and New Zealand, followed by North America and Northern and Western Europe. An average yearly increase of more than four percent has occurred globally for the past twenty years [4]. Intermittent sun exposure (characterized by a history of sunburns along with a weakened immune system), a family history, and prior removal of melanomas are considered significant risk factors [3][5][6]. Melanoma accounts for about 1–4% of all skin cancers globally. Although it is less frequent, it is much deadlier than other skin tumors [6].
2. Melanocyte Function and the Emergence of Melanoma
Within the melanocytes are melanosomes that produce the pigment melanin [7]. In response to UV-induced DNA base damage, keratinocytes in the skin produce a melanocyte-stimulating hormone (αMSH) that binds to the melanocortin receptor 1 (MC1R) on melanocytes, ultimately leading to the activation of microphthalmia-associated transcription factor (MITF) (Figure 1). MITF upregulates the expression of enzymes required for melanogenesis and is considered the main regulator of melanocyte differentiation. Amplification in MITF is present in about 20% of melanomas and is associated with reduced five-year survival [7]. Melanin and the distribution of melanosomes in the epidermis are among the most critical factors in protecting human skin from the harmful effects of UV radiation [8][9]. UVB (280–315 nm) radiation is considered a primary mutagen because it is absorbed directly by DNA, inducing DNA base damage. On the other hand, UVA (315–400 nm) is mainly responsible for indirect DNA damage by generating reactive oxygen species (ROS) [10][11].
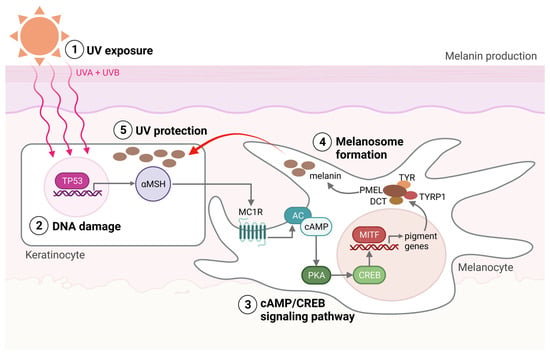
Figure 1. Melanin production. (1) The production of melanin, a process called melanogenesis, is activated upon UV exposure. (2) UV radiation damages the DNA in the keratinocytes, activating the TP53 pathway, which results in the production of αMSH. αMSH is secreted from the keratinocytes and binds to the MC1R on the melanocyte. (3) cAMP levels are then increased within the melanocytes, activating protein kinase A (PKA). PKA activation induces the recruitment of CRE-binding (CREB) protein and thereby the transcriptional activity of MITF. (4) MITF activates the transcription of pigment genes, including TYR, TYRP1, DCT, and PMEL, which are transported to the membrane-bound melanosome. (5) Matured melanosomes are then transferred from melanocytes to keratinocytes to protect them against UV light. Created with BioRender.com (accessed on 19 September 2023).
3. Melanoma Diagnosis
The preferred method to diagnose melanoma is surgical excision, with appropriate margins, followed by a histological examination of the skin lesion. Parameters such as histological subtype, number of mitoses, and Breslow thickness are determined, as well as the presence or absence of ulceration and microsatellites. Additional information regarding the growth phase, the presence or absence of tumor-infiltrating lymphocytes (TILs), signs of regression, and vascular or perineural involvement may also be assessed. If there are uncertainties regarding the diagnosis, immunohistochemical analysis provides tools to differentiate malignant lesions from benign nevi. The melanoma markers MLANA (MART-1), TYR, HMB45 (PMEL), SOX10, S100, and the proliferation marker MIB-1, which recognizes the Ki67 antigen, are used to diagnose primary and metastatic melanoma. Using these markers, melanoma can be distinguished from other cancer types such as epithelial tumors, neuroendocrine tumors, sarcomas, lymphomas, and germ cell tumors [12][13][14][15][16][17][18].
4. Melanoma Susceptibility
About 10% of melanomas occur in patients with a family history of melanoma [19][20]. Although most genetic alterations associated with melanoma development are somatic, the underlying presence of heritable melanoma risk genes is essential to disease occurrence. This is believed to be the case even in “sporadic” melanoma [21][22]. In the familial atypical multiple mole-melanoma (FAMMM) syndromes and the melanoma–astrocytoma syndrome (MAS), germline mutations in CDKN2A and CDK4 are the most frequent genetic abnormalities [20]. CDKN2A encodes two crucial tumor suppressor proteins, p16INK4A, and p14ARF, which, together with CDK4, function as cell cycle regulators [23][24].
Polymorphism of the MC1R gene has been found to play a significant role in sporadic melanoma of the skin. MC1R, the key regulator of skin pigmentation, has more than 200 coding region variants identified, many within the European population [25]. Polymorphisms in the MC1R gene give rise to diverse skin pigmentation phenotypes, among which red hair, freckles, and fair skin express low pigmentation. Increased vulnerability to melanoma is a consequence of a quantitative shift of melanin synthesis from eumelanin to pheomelanin [25]. Furthermore, specific variants of MC1R are believed to increase the penetrance of CDKN2A mutations, doubling the risk of melanoma compared to a CDKN2A mutation alone.
5. Dysregulated Signaling Pathways in Melanoma
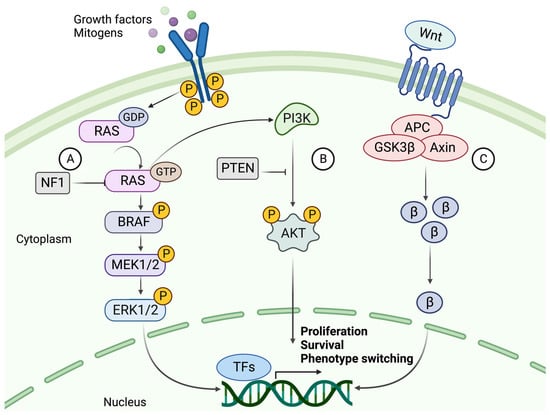
The mutation rate in melanoma tumors, measured as the number of mutations per Mb, exceeds that of all other cancers according to The Cancer Genome Atlas (TCGA) data [2][26]. The high rate of somatic mutations makes it difficult to distinguish between causative and bystander mutations. The major players involved in melanoma formation include cell-autonomous mutations of the MAPK, PI3K, and WNT signaling pathways (Figure 2) [5][27].
Figure 2. The signaling cascades of MAPK, PI3K, and canonical WNT pathways. (A) The NRAS-BRAF MAPK pathway is activated by binding a growth factor (GF) or mitogen to an RTK. Upon activation, the RAS protein phosphorylates (P) MEK1/2, phosphorylating ERK1/2. ERK can then translocate to the nucleus and activate transcription factors which promote proliferation and progression through the cell cycle. NF1 hampers this cell cycle progression by converting RAS to its inactive GDP-bound form. (B) The activation of PI3K signaling by GTP-bound RAS. PI3K activates AKT through phosphorylation using a second messenger (PIP3 not shown). AKT is a kinase that mediates the phosphorylation of protein substrates, subsequently affecting the cell cycle and survival of the tumor cell. PTEN acts as a suppressor of this pathway by dephosphorylating PIP3, thereby blocking the activation of AKT. (C) The canonical WNT signaling pathway and its main effector, β-catenin (β). β-Catenin is activated upon binding a WNT ligand to the G protein-coupled receptor (GPCR), whereby it is prevented from degradation and instead accumulates in the cytoplasm. β-Catenin then relocates to the nucleus, where it acts as a co-activator of transcription of genes such as those related to EMT-like phenotype switching. Created with BioRender.com (accessed on 19 September 2023).
5.1. The Mitogen-Activated Protein Kinase (MAPK) Pathway
The most common mutations in melanoma involve the MAPK signaling pathway, where acquired mutations in the genes encoding two kinases, BRAF and NRAS, result in continuous activation and aberrant cell proliferation. Constant activation of MAPK signaling leads to increased expression of cyclin D1, which interacts with CDK4/6 to promote phosphorylation and inhibition of the retinoblastoma (Rb) family of transcriptional repressors, enhancing E2F-dependent transcription of S-phase genes, facilitating G1-S phase transition and consequently cell cycle progression [28]. Upon acquirement of a BRAF mutation, a benign melanocytic lesion does not switch to malignancy.
BRAF mutations are found in about half of all melanocytic lesions and are thought to occur early in the disease. Most mutations occur at the V600 position, where a valine-to-glutamate substitution (V600E) is the most common (~80–90%). V600K, V600D, and V600R account for another 10% to 15% of mutations. BRAF mutations are more frequent in melanomas that develop in sun-exposed skin. The mutated BRAF kinase can activate its downstream effector MEK, independent of RAS activation. MEK then activates ERK, which relocates to the nucleus and alters gene transcription by increasing the expression of various transcription factors, such as c-MYC and MITF (Figure 2) [29][30].
NRAS and BRAF mutations are almost always mutually exclusive, where NRAS mutations occur in about 20% of melanoma cases. In addition to MAPK pathway activation, oncogenic RAS acts as a positive upstream regulator of the PI3K pathway [31]. Thus, activation of RAS transcription leads to downstream activation of two interconnected pathways.
5.2. The PI3K/AKT Signaling Pathway
To overcome OIS in a BRAF-mutated melanocytic lesion, subsequent mutations of the PI3K pathway are frequently acquired [32]. The coexistence of mutations in these pathways has been shown to overcome BRAFV600E OIS by the loss of PTEN expression or by overexpression of AKT3, a downstream target of PI3K (Figure 2). PTEN-negative or AKT3-overexpressing melanomas do not undergo apoptosis in response to BRAF inhibition [33]. PTEN is also thought to inhibit MAPK signaling by decreasing the phosphorylation of MEK and ERK [34].
5.3. Canonical and Noncanonical WNT Signaling
Normal WNT signaling is required for melanocyte development, while aberrant WNT signaling is known to contribute to melanoma formation [27]. The canonical WNT pathway mainly contributes to the development of primary melanoma by suppressing p16INK4A, overcoming OIS, and increasing proliferation. By contrast, the noncanonical pathway is primarily involved in metastasis formation by disrupting cell polarity and increasing migration capabilities [35].
5.4. The Role of KIT, NF1, TERT, and TP53 in Melanoma
Other essential effector molecules in melanoma formation include KIT, NF1, TERT, and p53. KIT is a proto-oncogene that encodes a receptor tyrosine kinase (RTK) and is considered a driver mutation in melanoma. It is found to be mutated in about 1–3% of melanoma tumors [36]. KIT activates the MAPK and PI3K pathways leading to cell proliferation and survival. NF1 is a tumor suppressor that negatively regulates the MAPK and PI3K pathways. NF1 mutations are prevalent in melanomas that are wild-type for BRAF and NRAS, NF1 are considered driver mutations in these patients [37].
6. The Tumor Microenvironment
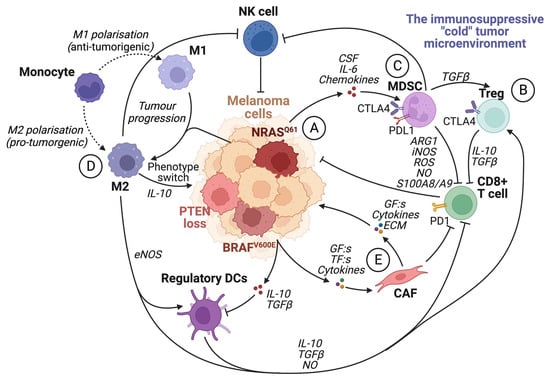
The tumor microenvironment (TME) is known to have a central role in cancer development. A pro-tumorigenic microenvironment is essential for a tumor’s survival and progression [38]. Compared to the microenvironment surrounding normal tissue, the TME differs in its extracellular matrix (ECM) composition, metabolism, nutritional status, pH, and oxygen levels [39]. The TME consists of a complex mixture of fibroblasts, immune cells, and endothelial cells embedded in the ECM. There is continuous reciprocal communication between tumor cells and the microenvironment, where tumor cells secrete stimulatory growth factors, chemokines, and cytokines, as well as induce changes in glucose and oxygen levels [27][38][39]. This results in the recruitment of stromal cells, immune cells, and vascular cells, which remodels the surroundings and creates a pro-tumorigenic microenvironment (Figure 3).
Figure 3. The immunosuppressive tumor microenvironment. The recruitment of various cells modulates the TME by the secretion of cytokines and chemokines by tumor cells and other infiltrating cells. Most cells within an immunosuppressive tumor microenvironment inhibit the activation and function of cytotoxic CD8+ T cells. (A) Tumor cells can induce the activity of Treg cells, tumor-associated macrophages (M2), and MDSCs by secreting growth factors such as VEGF. They also facilitate the transformation of fibroblasts into cancer-associated fibroblasts and enhance the expression of PD1 on CD8+ T cells. (B) Treg cells inhibit CD8+ T cells and NK cells by upregulating CTLA4 and releasing IL-10 and TGFβ. (C) MDSC expression of PDL1 inhibits T-cell activation by binding to PD1. Furthermore, MDSCs promote Treg cell proliferation in a TGFβ-dependent manner, boost angiogenesis in the tumor microenvironment, and contribute to the phenotypic switch in melanoma cells. In addition, MDSCs hinder CD8+ T cells by releasing arginase I and S100A8/A9 and metabolites such as ROS, NO, and iNOS. (D) TAMs promote regulatory DC maturation, inhibition of CD8+ T cells and NK cells, and facilitate phenotype switching in melanoma cells by IL-10 signaling. (E) Cancer-associated fibroblasts (CAFs) can induce immunosuppression by inhibiting CD8+ T cells. CAFs also secrete TGFβ, CXCL12, matrix metalloproteinase 2 (MMP2), and IL-6, which promote tumor proliferation and invasion. Created with BioRender.com (accessed on 19 September 2023).
6.1. Metabolic Reprogramming of the Tumor Microenvironment
Oncogenic reprogramming of cellular metabolism is an essential feature of melanoma progression. It is triggered by genetic alterations and adaptations, forming a microenvironment that lacks nutrients and oxygen [39][40][41]. Metabolic reprogramming from oxidative phosphorylation (OXPHOS) to a glycolytic phenotype has long been considered a hallmark of cancer [42]. Indeed, a key feature of BRAFV600E mutated melanoma is the metabolic reprogramming from mitochondrial respiration to glycolysis [41]. However, there is increasing evidence linking mitochondrial pathways to cancer development and progression [43][44][45][46]. Mitochondrial metabolism impacts tumor development by increasing ROS as a byproduct of OXPHOS, supporting the genomic instability required for transforming a melanocytic lesion into a melanoma tumor [41].
6.2. Phenotype Switching of Melanoma Cells
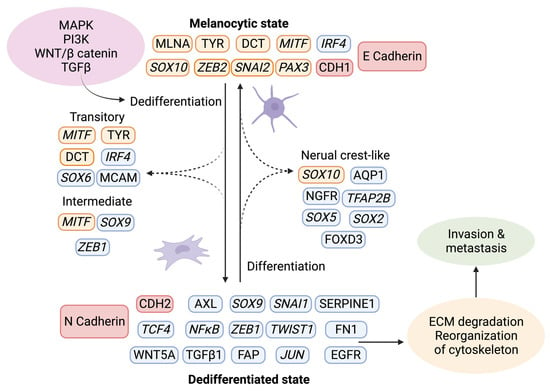
The tumor stroma further contributes to melanoma progression by initiating a “phenotypic switch”, whereby the melanocytic characteristics are lost and exchanged in favor of a more dedifferentiated phenotype (Figure 4) [47]. The plasticity of melanoma cells shares characteristics of neuroendocrine tumors [48], germ cell cancers [49][50], and epithelial tumors. The dedifferentiated mesenchymal-like melanoma cells almost completely lose their melanocytic features and appear nearly undistinguishable from epithelial tumors undergoing EMT [51][52][53].
Figure 4. Phenotype switching in melanoma. Upon activation of the signaling pathways MAPK, PI3K, WNT, and TGFβ, a changed expression of specific markers leads to ECM degradation and reorganization of the cytoskeleton, which facilitates invasion and metastasis. Using single-cell approaches, it has become evident that phenotype switching is not binary but rather a multistep process in which cells transit through a series of intermediate cell states expressing combinations of epithelial and mesenchymal phenotypes. These intermediate states can simultaneously display proliferation, invasion, and stemness features. Transcriptional regulators are highlighted in italics. The red indicates the switch between E- and N-cadherin and the responsible genes CDH1 and CDH2. Melanocytic markers are highlighted in orange, and other markers are in blue. Created with BioRender.com (accessed on 19 September 2023).Given that melanocytes derive from neural crest cells, which underwent EMT during their development into melanocytes, it is not surprising that phenotype switching mimics this process [54]. The phenotypic transition of melanoma cells includes the acquisition of a spindle-like morphology, upregulation of mesenchymal markers, and release of ECM degrading proteins, such as matrix metalloproteinases (MMPs) which further support invasiveness. An additional downregulation of the epithelial cell surface protein E-cadherin in favor of N-cadherin accompanies the phenotypic switch. As a result, the keratinocytes in the skin lose control over the melanoma cells, which gain properties to grow vertically through the epidermis [55]. A parallel upregulation of TGFβ signaling and the above-described pathways (MAPK, PI3K/AKT3, and Wnt/β-catenin) increase the abundance of transcription factors such as MITF, SOX, Snai1/2, TCF4, Twist1, Zeb1/2, PAX3, and NFκB, further facilitating phenotype switching [56][57][58]. Melanoma is believed to progress and metastasize by alternating between the melanocytic/proliferative and dedifferentiated/invasive states, including intermediate (proliferative and invasive), transitory (proliferative and invasive), and neural crest stem cell-like (NCSC-like) states (invasive) [47][59].
7. The Immune System
7.1. The Roles of the Different Immune Cells in Melanoma Development
Melanoma tumors are considered immunogenic, and often, they have a higher infiltration of immune cells compared to other cancer types. These immune cells can have opposing effects, either initiating or inhibiting the immune response [60]. Macrophages constitute a large pool of immune cells in solid tumors, and other cell types include NK cells, lymphocytes, dendritic cells (DCs), mast cells, and neutrophils [39][60]. Macrophages present within a tumor can have very different functions, which are divided into two main groups: pro-tumorigenic (M2) and antitumorigenic (M1) (Figure 3). The M1 macrophages are often more active in the earlier stages of tumor formation and exert immune activation, apoptosis, and inflammatory functions. On the other hand, the M2 macrophages are often the most dominant in the later disease stages. The M2 macrophages promote tumor growth, angiogenesis, invasiveness, and immune suppression. Macrophages utilizing tumor-suppressive functions are often called tumor-associated macrophages (TAMs). TAMs can switch between M1 and M2, depending on the stimulus from the surrounding tissue [39].
7.2. The Spatial Architecture of the Tumor Immune Microenvironment
The composition and molecular properties of immune cells in the tumor and TME are well-studied [61][62][63][64][65]. More recently, the spatial architecture of these cells has received increased attention to elucidate the varying treatment responses to ICIs [66][67]. Technologies including multiplexed IHC and immunofluorescent (IF) imaging [68][69], mass cytometry [70], and single-cell multiomics [67][71] have been developed for the in-depth study of immune cell phenotypes, their spatial patterns, and their interactions with other cells.
Single marker IHC has long been used to study immune cells in melanoma, mainly focusing on their density and distribution and how that impacts prognosis prediction [72][73][74]. Melanomas containing a high number of CD8+ T cells in the stromal compartment and within the tumor parenchyma are considered “hot”, while “cold” melanomas are characterized by a scarce immune infiltrate [75][76]. The use of PD-L1 expression alone to predict treatment response has shortcomings [77].
Multiplexed imaging techniques, such as brightfield (BF) multiplexed IHC, can visualize up to eight markers simultaneously by labeling different cell populations on the same section [78]. Immune cell markers including CD3 (T cell activation), CD8 (cytotoxic T cells), CD20 (B cells), CD68 (macrophages), CD163 (M2 macrophages), and CD16 (cytotoxic macrophages and NK cells) can then be combined with PD-L1, SOX10, and Ki67 [68][79]. Thus, simultaneous determination of the location and interaction between immune cell subpopulations and melanoma cells can be accomplished.
8. Molecular Classification of Melanoma—A Way Forward
In addition to the clinical and pathologic classifications of melanoma [14][80][81][82][83], there is increasing interest in molecular classifications that aim to stratify patients into clinically meaningful subgroups to guide treatment selection, prognosis prediction, and patient outcomes.
In 2006, Hoek et al. outlined a transcriptional taxonomy for melanoma, using cell lines based on gene expression profiling [59]. They proposed three groups with different metastatic potentials. Subclasses A and B were proliferative with weak metastatic potential and displayed an NCSC-like transcriptomic signature, while subclass C was less proliferative but with high metastatic potential. The subtypes were primarily driven by WNT- and TGFβ-like signaling. In 2010, Jönsson et al. proposed a molecular stratification of metastatic melanoma samples based on gene expression profiling. Tumors were divided into four distinct subtypes “high-immune”, “proliferative”, “pigmentation”, and “normal-like”, as reflected by their characteristic gene expression [84].
9. The Relationship between Molecular Mechanisms and Treatment Response
9.1. Immunotherapy
Melanoma tumors are considered one of the most immunogenic tumors with a high mutational burden and are therefore well suited for immunotherapy [85][86]. Indeed, melanoma was the first cancer type treated with immune checkpoint therapy [87]. There are currently four approved targets, including CTLA4, PD1, PD-L1, and LAG-3. These proteins are expressed on the surface of T cells, among other cell types, and are involved in signaling pathways that lead to immune suppression. LAG-3 negatively regulates CD4+ T-cell activation and function while enhancing Treg activity and was the most recently approved immune checkpoint target [88]. LAG-3 can act synergistically with PD1 targets [89]. The approved immune checkpoint inhibitors exert their function by binding to these proteins and blocking their activity, thereby reestablishing an antitumorigenic immune environment [90].
9.2. Targeted Therapy
Approximately 70% of melanoma patients harbor a genetic alteration in one of the main signaling pathways previously described. The MAPK signaling pathway consists of an RTK and the proteins RAS, RAF, MEK, and ERK. Small molecule inhibitors of BRAF and MEK are approved as targets for melanoma therapy (MAPKi) [35]. However, resistance to these inhibitors is an immense problem. When used in combination, increased efficacy combined with reduced toxicity is observed, albeit long-term responses to targeted therapies are rare [90]. In response to MAPKi therapy, melanoma cells often downregulate MITF and upregulate RTKs including AXL, EGFR, and PDGFRβ, resulting in a dedifferentiated MITFlow/AXLhigh phenotype [91][92].
9.3. Novel Treatments
New promising therapies to combat melanoma include cancer vaccines based on predicted neoantigens. Due to the high mutation rate in melanoma, the mutational landscape does not overlap between patients. Cancer vaccines induce T-cell reactivity based on the genome of a particular tumor by predicting potential neoantigens [89][93]. However, despite T-cell reactivity being induced, the long-term efficacy still relies on the continuous activation of these cells, which a suppressive TME might hamper. Therefore, combining cancer vaccines with immune checkpoint inhibitors may produce a more efficient therapy response.
Another approach includes nanosystems that aim to improve drug efficacy through personalized and targeted drug delivery. By associating a melanoma treatment, such as immune checkpoint inhibitors or targeted therapies, with a nanoparticle delivery system, the drug can be protected from degradation, and the harmful effects on healthy cells can be minimized [15][94][95].
10. Conclusions
Melanoma and many other cancers represent major public health problems. Disease management of melanoma is challenging due to its heterogeneous nature and unpredictable pattern of progression. The emergence of immunotherapy and targeted therapy has prolonged numerous lives, but many patients relapse or do not respond to treatment. Although resistance to immunotherapies may manifest at different times, similar or overlapping mechanisms are often seen [60][96][97]. As outlined, melanoma tumors display a complex landscape with subpopulations of clones with different mutations surrounded by a constantly changing TME. As a result, exceptional intratumoral heterogeneity is observed in both primary tumors and metastases from melanoma patients.
References
- Lallier, T.E. Cell lineage and cell migration in the neural crest. Ann. N. Y. Acad. Sci. 1991, 615, 158–171.
- Grzywa, T.M.; Paskal, W.; Wlodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Coricovac, D.; Dehelean, C.; Moaca, E.A.; Pinzaru, I.; Bratu, T.; Navolan, D.; Boruga, O. Cutaneous Melanoma-A Long Road from Experimental Models to Clinical Outcome: A Review. Int. J. Mol. Sci. 2018, 19, 1566.
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503.
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65.
- Naik, P.P. Cutaneous Malignant Melanoma: A Review of Early Diagnosis and Management. World J. Oncol. 2021, 12, 7–19.
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122.
- Kollias, N.; Sayre, R.M.; Zeise, L.; Chedekel, M.R. Photoprotection by melanin. J. Photochem. Photobiol. B Biol. 1991, 9, 135–160.
- Raposo, G.; Marks, M.S. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797.
- Young, A.R.; Potten, C.S.; Nikaido, O.; Parsons, P.G.; Boenders, J.; Ramsden, J.M.; Chadwick, C.A. Human melanocytes and keratinocytes exposed to UVB or UVA in vivo show comparable levels of thymine dimers. J. Investig. Dermatol. 1998, 111, 936–940.
- Fadadu, R.P.; Wei, M.L. Ultraviolet A radiation exposure and melanoma: A review. Melanoma Res. 2022, 32, 405–410.
- Țăpoi, D.A.; Gheorghișan-Gălățeanu, A.-A.; Dumitru, A.V.; Ciongariu, A.M.; Furtunescu, A.R.; Marin, A.; Costache, M. Primary Undifferentiated/Dedifferentiated Cutaneous Melanomas—A Review on Histological, Immunohistochemical, and Molecular Features with Emphasis on Prognosis and Treatment. Int. J. Mol. Sci. 2023, 24, 9985.
- Gheoca Mutu, D.E.; Avino, A.; Balcangiu-Stroescu, A.E.; Mehedințu, M.; Bălan, D.G.; Brîndușe, L.A.; Popescu, A.M.; Ionescu, D.; Cristea, B.M.; Tomescu, L.F.; et al. Histopathological evaluation of cutaneous malignant melanoma: A retrospective study. Exp. Ther. Med. 2022, 23, 402.
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255.
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652.
- Weinstein, D.; Leininger, J.; Hamby, C.; Safai, B. Diagnostic and prognostic biomarkers in melanoma. J. Clin. Aesthetic Dermatol. 2014, 7, 13–24.
- Banerjee, S.S.; Harris, M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology 2000, 36, 387–402.
- Nwafor, J.N.; Torere, B.E.; Agu, E.; Kadiku, L.; Ogunyemi, T.; Akinsanya, P.A.; Araromi, O.O.; Akahara, D.E.; Okobi, O.E. The Role of Biomarkers in the Diagnosis and Prognosis of Different Stages of Melanoma. Cureus 2023, 15, e38693.
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080.
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Dermatol. 2016, 74, 395–407; quiz 408–410.
- Lu, Y.; Ek, W.E.; Whiteman, D.; Vaughan, T.L.; Spurdle, A.B.; Easton, D.F.; Pharoah, P.D.; Thompson, D.J.; Dunning, A.M.; Hayward, N.K.; et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum. Mol. Genet. 2014, 23, 6112–6118.
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14.
- Mukherjee, B.; Delancey, J.O.; Raskin, L.; Everett, J.; Jeter, J.; Begg, C.B.; Orlow, I.; Berwick, M.; Armstrong, B.K.; Kricker, A.; et al. Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J. Natl. Cancer Inst. 2012, 104, 953–956.
- Bandarchi, B.; Ma, L.; Navab, R.; Seth, A.; Rasty, G. From melanocyte to metastatic malignant melanoma. Dermatol. Res. Pract. 2010, 2010, 583748.
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018, 10, 1143–1154.
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218.
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784.
- Bartek, J.; Lukas, J. Cyclin D1 multitasks. Nature 2011, 474, 171–172.
- Zuber, J.; Tchernitsa, O.I.; Hinzmann, B.; Schmitz, A.C.; Grips, M.; Hellriegel, M.; Sers, C.; Rosenthal, A.; Schäfer, R. A genome-wide survey of RAS transformation targets. Nat. Genet. 2000, 24, 144–152.
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290.
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114.
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724.
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008, 68, 3429–3439.
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552.
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 424.
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571.
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263.
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515.
- Kimm, M.A.; Klenk, C.; Alunni-Fabbroni, M.; Kästle, S.; Stechele, M.; Ricke, J.; Eisenblätter, M.; Wildgruber, M. Tumor-Associated Macrophages-Implications for Molecular Oncology and Imaging. Biomedicines 2021, 9, 374.
- Neagu, M. Metabolic Traits in Cutaneous Melanoma. Front. Oncol. 2020, 10, 851.
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572.
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280.
- Roberts, E.R.; Thomas, K.J. The role of mitochondria in the development and progression of lung cancer. Comput. Struct. Biotechnol. J. 2013, 6, e201303019.
- Zhang, X.; Tai, Z.; Miao, F.; Huang, H.; Zhu, Q.; Bao, L.; Chen, Z. Metabolism heterogeneity in melanoma fuels deactivation of immunotherapy: Predict before protect. Front. Oncol. 2022, 12, 1046102.
- Pedri, D.; Karras, P.; Landeloos, E.; Marine, J.C.; Rambow, F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022, 289, 1352–1368.
- Cham, J.; Shavit, A.; Ebrahimi, A.; Viray, M.; Gibbs, P.; Bhangoo, M.S. Malignant Melanoma With Neuroendocrine Differentiation: A Case Report and Literature Review. Front. Oncol. 2021, 11, 763992.
- Ricci, C.; Franceschini, T.; Giunchi, F.; Grillini, M.; Ambrosi, F.; Massari, F.; Mollica, V.; Colecchia, M.; Fiorentino, M. Immunohistochemical Expression of Preferentially Expressed Antigen in Melanoma (PRAME) in the Uninvolved Background Testis, Germ Cell Neoplasia In Situ, and Germ Cell Tumors of the Testis. Am. J. Clin. Pathol. 2022, 157, 644–648.
- Nettersheim, D.; Schorle, H. The plasticity of germ cell cancers and its dependence on the cellular microenvironment. J. Cell. Mol. Med. 2017, 21, 1463–1467.
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759.
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318.
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683.
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 1387.
- Hoek, K.; Rimm, D.L.; Williams, K.R.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H.M.; Berger, A.J.; Cheng, E.; Trombetta, E.S.; et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004, 64, 5270–5282.
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140.
- Hodorogea, A.; Calinescu, A.; Antohe, M.; Balaban, M.; Nedelcu, R.I.; Turcu, G.; Ion, D.A.; Badarau, I.A.; Popescu, C.M.; Popescu, R.; et al. Epithelial-Mesenchymal Transition in Skin Cancers: A Review. Anal. Cell. Pathol. 2019, 2019, 3851576.
- Sanna, A.; Phung, B.; Mitra, S.; Lauss, M.; Choi, J.; Zhang, T.; Njauw, C.N.; Cordero, E.; Harbst, K.; Rosengren, F.; et al. DNA promoter hypermethylation of melanocyte lineage genes determines melanoma phenotype. JCI Insight 2022, 7, e156577.
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302.
- Kalaora, S.; Nagler, A.; Wargo, J.A.; Samuels, Y. Mechanisms of immune activation and regulation: Lessons from melanoma. Nat. Rev. Cancer 2022, 22, 195–207.
- Chan, M.K.-K.; Chung, J.Y.-F.; Tang, P.C.-T.; Chan, A.S.-W.; Ho, J.Y.-Y.; Lin, T.P.-T.; Chen, J.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; et al. TGF-β signaling networks in the tumor microenvironment. Cancer Lett. 2022, 550, 215925.
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325.
- Gunaydin, G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349.
- Grisaru-Tal, S.; Rothenberg, M.E.; Munitz, A. Eosinophil–lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat. Immunol. 2022, 23, 1309–1316.
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102.
- Fu, T.; Dai, L.-J.; Wu, S.-Y.; Xiao, Y.; Ma, D.; Jiang, Y.-Z.; Shao, Z.-M. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 2021, 14, 98.
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24.
- Bravo, A.I.; Aris, M.; Panouillot, M.; Porto, M.; Dieu-Nosjean, M.C.; Teillaud, J.L.; Barrio, M.M.; Mordoh, J. HEV-associated dendritic cells are observed in metastatic tumor-draining lymph nodes of cutaneous melanoma patients with longer distant metastasis-free survival after adjuvant immunotherapy. Front. Immunol. 2023, 14, 1231734.
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217.
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422.
- Hsieh, W.-C.; Budiarto, B.R.; Wang, Y.-F.; Lin, C.-Y.; Gwo, M.-C.; So, D.K.; Tzeng, Y.-S.; Chen, S.-Y. Spatial multi-omics analyses of the tumor immune microenvironment. J. Biomed. Sci. 2022, 29, 96.
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080.
- Koh, J.; Kwak, Y.; Kim, J.; Kim, W.H. High-Throughput Multiplex Immunohistochemical Imaging of the Tumor and Its Microenvironment. Cancer Res. Treat. 2019, 52, 98–108.
- Capelozzi, V.L.; Parra, E.R. Editorial: Multiplexed image analysis for translational research project applications. Front. Oncol. 2022, 12, 1089265.
- Sobottka, B.; Nowak, M.; Frei, A.L.; Haberecker, M.; Merki, S.; Levesque, M.P.; Dummer, R.; Moch, H.; Koelzer, V.H. Establishing standardized immune phenotyping of metastatic melanoma by digital pathology. Lab. Investig. A J. Tech. Methods Pathol. 2021, 101, 1561–1570.
- Attrill, G.H.; Ferguson, P.M.; Palendira, U.; Long, G.V.; Wilmott, J.S.; Scolyer, R.A. The tumour immune landscape and its implications in cutaneous melanoma. Pigment Cell Melanoma Res. 2021, 34, 529–549.
- Kuczkiewicz-Siemion, O.; Sokół, K.; Puton, B.; Borkowska, A.; Szumera-Ciećkiewicz, A. The Role of Pathology-Based Methods in Qualitative and Quantitative Approaches to Cancer Immunotherapy. Cancers 2022, 14, 119.
- Sun, Z.; Nyberg, R.; Wu, Y.; Bernard, B.; Redmond, W.L. Developing an enhanced 7-color multiplex IHC protocol to dissect immune infiltration in human cancers. PLoS ONE 2021, 16, e0247238.
- Ugolini, F.; Pasqualini, E.; Simi, S.; Baroni, G.; Massi, D. Bright-Field Multiplex Immunohistochemistry Assay for Tumor Microenvironment Evaluation in Melanoma Tissues. Cancers 2022, 14, 3682.
- Situm, M.; Buljan, M.; Kolić, M.; Vučić, M. Melanoma—Clinical, dermatoscopical, and histopathological morphological characteristics. Acta Dermatovenerol. Croat. 2014, 22, 1–12.
- Duncan, L.M. The classification of cutaneous melanoma. Hematol. Oncol. Clin. N. Am. 2009, 23, 501–513.
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784.
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99.
- Jonsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringner, M.; Lillehaug, J.R.; Borg, A.; Lonning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367.
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment-Update 2022. Eur. J. Cancer 2022, 170, 256–284.
- Hodi, F.S.; Chiarion-Sileni, V.; Lewis, K.D.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-term survival in advanced melanoma for patients treated with nivolumab plus ipilimumab in CheckMate 067. J. Clin. Oncol. 2022, 40, 9522.
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297.
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34.
- Ni, J.-j.; Zhang, Z.-z.; Ge, M.-j.; Chen, J.-y.; Zhuo, W. Immune-based combination therapy to convert immunologically cold tumors into hot tumors: An update and new insights. Acta Pharmacol. Sin. 2022, 44, 288–307.
- Lee, K.A.; Nathan, P. Cutaneous Melanoma-A Review of Systemic Therapies. Acta Derm. Venereol. 2020, 100, adv00141.
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977.
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712.
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221.
- Beiu, C.; Giurcaneanu, C.; Grumezescu, A.M.; Holban, A.M.; Popa, L.G.; Mihai, M.M. Nanosystems for Improved Targeted Therapies in Melanoma. J. Clin. Med. 2020, 9, 318.
- Verma, J.; Warsame, C.; Seenivasagam, R.K.; Katiyar, N.K.; Aleem, E.; Goel, S. Nanoparticle-mediated cancer cell therapy: Basic science to clinical applications. Cancer Metastasis Rev. 2023, 1–27.
- Huang, F.; Santinon, F.; Flores González, R.E.; Del Rincón, S.V. Melanoma Plasticity: Promoter of Metastasis and Resistance to Therapy. Front. Oncol. 2021, 11, 756001.
- Pagliuca, C.; Di Leo, L.; De Zio, D. New Insights into the Phenotype Switching of Melanoma. Cancers 2022, 14, 6118.
More
Information
Subjects:
Cell Biology; Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
486
Revisions:
3 times
(View History)
Update Date:
06 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No