Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rabindra Roy | -- | 3296 | 2023-09-25 13:56:56 | | | |
| 2 | Camila Xu | Meta information modification | 3296 | 2023-09-26 04:10:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gohil, D.; Sarker, A.H.; Roy, R. Base Excision Repair Mechanisms. Encyclopedia. Available online: https://encyclopedia.pub/entry/49601 (accessed on 25 July 2026).
Gohil D, Sarker AH, Roy R. Base Excision Repair Mechanisms. Encyclopedia. Available at: https://encyclopedia.pub/entry/49601. Accessed July 25, 2026.
Gohil, Dhara, Altaf H. Sarker, Rabindra Roy. "Base Excision Repair Mechanisms" Encyclopedia, https://encyclopedia.pub/entry/49601 (accessed July 25, 2026).
Gohil, D., Sarker, A.H., & Roy, R. (2023, September 25). Base Excision Repair Mechanisms. In Encyclopedia. https://encyclopedia.pub/entry/49601
Gohil, Dhara, et al. "Base Excision Repair Mechanisms." Encyclopedia. Web. 25 September, 2023.
Copy Citation
Base excision repair (BER) corrects forms of oxidative, deamination, alkylation, and abasic single-base damage that appear to have minimal effects on the helix. Since its discovery in 1974, the field has grown in several facets: mechanisms, biology and physiology, understanding deficiencies and human disease, and using BER genes as potential inhibitory targets to develop therapeutics.
base excision repair (BER)
5′-Gap
nucleotide incision repair (NIR)
transcription-associated
1. Global
Global repair mechanisms can be applied to any section of the genome. Specifically, base excision repair (BER) corrects forms of oxidative, deamination, alkylation, and abasic single-base damage that appear insignificant to the helix [1]. Oxidative damage, from either metabolic or environmental sources, can generate ROS, affecting both the DNA strand and dNTP pool. Oxidized bases can lead to abasic sites, and accumulation of such damage can result in mutagenesis and cell death [2]. Damage within the dNTP pool can result in the incorporation of oxidized bases during repair or replication, ultimately leading to ligation failure. Currently, three categories of DNA glycosylases recognize damaged bases and there are two repair classifications—short-nucleotide (SN-BER) or long-patch (LP-BER). Within global BER, nucleotide incision repair (NIR) is an alternate pathway. Figure 1 categorizes the DNA damage based on the DNA glycosylase-mediated repair intermediates and the six known repair pathways.
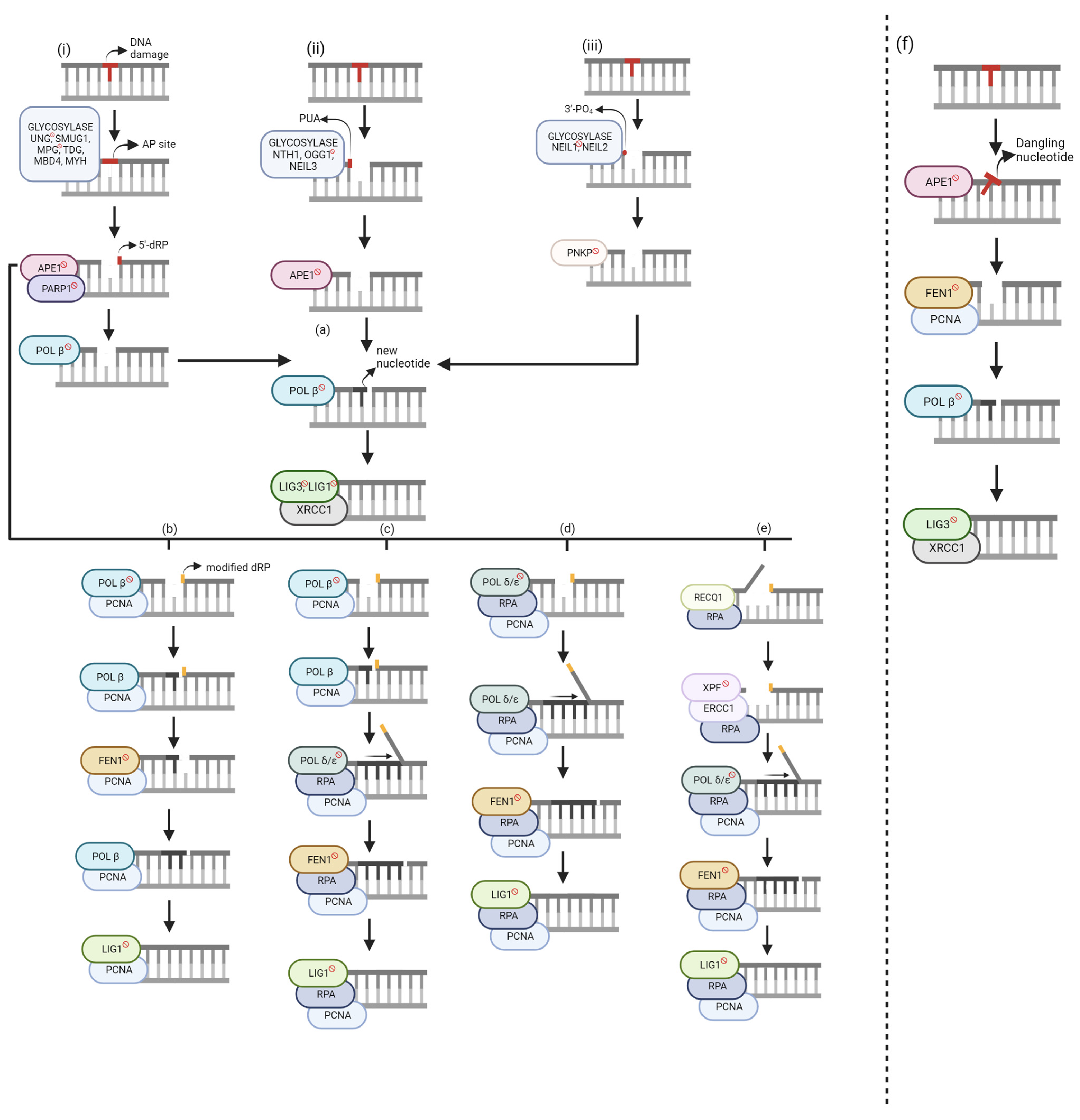
Figure 1. Global base excision repair (BER) mechanisms. Unlabeled DNA ends are either polymerase extendable clean 3′-OH groups or 5′-PO4 groups that can be ligated. (i) Monofunctional glycosylase. (ii) Bifunctional glycosylase—β elimination. (iii) Bifunctional glycosylase—β, δ-elimination. (a) Short-Nucleotide BER (SN-BER). (b–e) Long-Patch BER (LP-BER). (b) Hit and Run. (c) Polymerase Switch. (d) Non-Polymerase Switch. (e) 5′-Gap. (f) Short-Patch Nucleotide Incision Repair (NIR). Genes with [ ] sign are target BER inhibitors. The figure was generated using Biorender.com (accessed on 20 August 2023).
] sign are target BER inhibitors. The figure was generated using Biorender.com (accessed on 20 August 2023).
] sign are target BER inhibitors. The figure was generated using Biorender.com (accessed on 20 August 2023).Despite the BER pathway, various proteins are coordinated through transient protein–protein interactions or constitutive stable repair complexes [3]. The “passing the baton” model states that the repair intermediates are passed from one protein to the next until ligation is complete. However, its efficiency is limited to SN-BER. Another model suggests that the repair proteins are preassembled in a repair complex, as shown through co-immunoprecipitation experiments. Although there is no evidence of a stable complex containing all BER proteins in a single complex [3], research has shown that in addition to pre-existing sub-complexes, certain protein–protein interactions occur at the damage site [4][5][6][7][8]. In addition, glycosylase and APE1 may work independently of these complexes. Furthermore, it has been proposed that scaffolding accessory proteins may be needed for BER complex formation [9]. While it has no known catalytic function and is not required for in vitro BER reconstitution experiments, scaffolding proteins are needed for BER in vivo.
For efficient repair, the gene expression of every repair protein must be at its optimal level, which is assumed when discussing the various mechanisms in this section. The balance between SN- and LP-BER depends on the relative concentration of BER enzymes and scaffolding proteins and persistent 5′ blocking lesions at the repair site [10]. Genome instability, caused by over or under-expression of certain proteins, can lead to various deficiencies and diseases. An inefficient repair can lead to the accumulation of toxic lesions and, ultimately, apoptosis. For example, although base excision using a glycosylase is the first step in BER, higher amounts of these repair intermediates are more toxic than the parental damage if there are an insufficient amount of downstream repair proteins [11]. In addition, if a replication fork encounters a BER intermediate, the fork could collapse, resulting in a double-strand break [12], which can cause cell death if not repaired.
BER starts with a glycosylase excising the DNA base damage on the strand by hydrolyzing the N-glycosidic bond between the base and deoxyribose [13]. There are eleven known glycosylases, each recognizing specific DNA base lesions with overlapping specificities [14]. The choice of glycosylase for the same lesion may depend on the cellular state and cell types [15]. Glycosylases are categorized as either monofunctional or bifunctional. Monofunctional glycosylases (Figure 1i) only possess glycosylase activity and generate a hydrolytic, non-coding apurinic/apyrimidinic site (AP site) [16]. Conversely, bifunctional glycosylases have an additional function—3′-AP lyase activity. There are two groups of bifunctional glycosylases, which are segregated based on the type of reaction used to excise the DNA substrate, β-elimination (Figure 1ii) or β,δ-elimination (Figure 1iii), resulting in either 3′-phospho-α, β-unsaturated aldehyde (PUA), or a 3′-PO4, respectively.
AP sites and 3′-PUAs can be removed using human apurinic/apyrimidinic endonuclease 1 (APE1) but yield different products. APE1 works with poly-(ADP-ribose) polymerase 1 (PARP1) to remove the AP site by cleaving at the 5′ site, resulting in a 5′-deoxyribose phosphate (5′-dRP). When bound to DNA, PARP1 catalyzes poly(ADP-ribose) synthesis, allowing nuclear protein covalent modification. Conversely, autopoly(ADP-ribosyl)ation decreases DNA-binding activity and thus its dissociation from DNA. Although the mechanistic role of PARP1 is not entirely understood in BER, some functions have been explained. During POL β-mediated LP-BER stimulated by either APE1 or FEN1, PARP1 inhibited this pathway, but its effects were diminished once ribosylated [17]. There was no effect of PARP1 in SN-BER, impact on repair efficiency, or recruitment of BER proteins [3]. However, it protects single-strand breaks (SSBs) from nucleases or prevents the conversion to more toxic damage by dimerizing if the molar amount of SSBs exceeds the molar amount of needed BER enzymes [3]. APE1 removes the 3′-PUA, whereas polynucleotide kinase 3’-phosphatase (PNKP) excises 3′-PO4 groups, resulting in unmodified 3′ and 5′ ends. At this stage, either short-nucleotide or long-patch BER is chosen.
1.1. Short-Nucleotide BER (SN-BER)
Both monofunctional and bifunctional glycosylases can participate in SN-BER (Figure 1a). After a monofunctional glycosylase, APE1 generates a 5′-dRP, lysed by POL β’s lyase activity, resulting in unmodified 3′ and 5′ ends. From this point, all three glycosylase pathways have resulted in clean ends. Therefore, POL β can incorporate the correct nucleotide. The nick is sealed by DNA Ligase 3 (LIG3), with X-ray repair cross-complementing protein 1 (XRCC1) assisting. XRCC1 is a scaffold protein associated with POL β, LIG3, and human polynucleotide kinase (PNK), but its biochemical role is unknown [8]. Mutational analysis and binding studies have shown that LIG1 interacts with PCNA, and LIG3 interacts with the BRCT domain of XRCC1 [18]. Further research showed that LIG1 is applied to both SN-BER and LP-BER in nuclear DNA, while LIG3 repairs mitochondrial DNA. Still, it emphasized that further research must consider different base lesions, cell types, and other mammalian species [14]. Large amounts of damage to the dNTP pool can result in POL β incorporating an oxidized base, ultimately causing ligation failure [2]. The base correction function of BER is strongly backed up by trans-lesion DNA synthesis (TLS) polymerases; however, they often cause misincorporation and mutations [3].
1.2. Long-Patch BER (LP-BER)
Although SN-BER can be applied to any of the three pathways, LP-BER starts with a modified 5′-dRP. If the sugar group is either oxidized or reduced, POL β lyase activity cannot function, resulting in long-patch BER. Currently, there are four published LP-BER mechanisms with several commonalities. The primary differentiation factor between these sub-pathways is/are the polymerase(s) used for incorporating the new nucleotides.
Hit and Run
Although POL β cannot use its lyase activity to excise the modified 5′-dRP, a new mechanism within LP-BER showed that POL β and FEN1 work together using single-nucleotide gaps [19] (Figure 1b). The researchers showed that POL β had an increased binding affinity to the DNA substrate with excess FEN1 expression. This sub-pathway was discovered because it was revealed that although flaps with a 5′-terminal tetrahydrofuran (THF) residue were poor substrates for POL β-mediated repair, FEN1 stimulated DNA synthesis via POL β. In addition, PARP1 promoted POL β for LP-BER synthesis, but only in the presence of FEN1. However, the mechanism for this interaction is not known.
The researchers explained an alternating catalytic cycle between POL β and FEN1 for this sub-pathway. POL β incorporates one nucleotide, but the nicked flap remains, which is said to be the rate-limiting step. FEN1 cleaves the nicked flap and an additional nucleotide, creating a substrate for POL β to add another nucleotide via gap translation. While there would be no extra flaps, this alternating cycle would continue until ligation. This pathway needs proliferating cell nuclear antigen (PCNA) to localize the proteins to the DNA substrate and stimulate FEN1 activity [20][21]. Although initial research showed that this mechanism could be applied for a 2–11 nucleotide gap [19], further study segregated POL β-mediated LP-BER [17]. Gap translation can be used for 2-nucleotide repair. Alternatively, POL β can perform strand displacement when incorporating new nucleotides, allowing for an 11-nucleotide repair when stimulated by APE1 due to its 3′ to 5′ exonuclease activity [17]. POL β can perform strand displacement at high protein concentrations or when stimulated by specific protein–protein interactions [16]. Long-patch repair mechanisms use Ligase 1 (LIG1) rather than Ligase 3 (LIG3).
Polymerase Switch
POL β is involved in short-nucleotide and long-patch repair, but its highest catalytic activity is on 5′-phosphorylated 1-nucleotide (nt) gapped DNA [22]. Therefore, in this sub-pathway, POL β has been shown to incorporate the first nucleotide in the repair gap [23] (Figure 1c). POL δ/ε incorporates the remaining nucleotides to form the 2–11 nucleotide patch. POL δ/ε has 3′ to 5′ exonuclease activity, which is useful for proofreading errors created by proofreading-defective POL δ/ε molecules [24]. Whereas FEN1 cleaved 1-nt at a time in the Hit-and-Run mechanism, the 2–11 nt flap is now cleaved at once. Human replication protein A (RPA) stimulates FEN1 and LIG1 activity and binds to ssDNA for transient stability [25]. At the 5′ terminus, RPA binding allows for repair completion. Alternatively, RPA may bind at the 3′ end of the upstream primer to allow the DNA to remain unwound until POL ε can complete its proofreading function.
Non-Polymerase Switch
Unlike the previous sub-pathway, only POL δ/ε is needed for new nucleotide incorporation (Figure 1d). Although these polymerases can complete a 1-nt repair, the repair intermediate is inefficiently ligated [26] because a 1-nt incorporation does not generate strand displacement. Therefore, FEN1 would not be able to cleave the modified 5′-dRP. Downstream steps are like the Polymerase Switch sub-pathway.
5′-Gap
In 2017, an LP-BER sub-pathway was discovered that creates a 9-nt gap on the 5′ side (Figure 1e) [27]. RECQ1, a DNA helicase, unwinds the DNA and due to its 3′–5′ activity, creates an 8-nt long 5′-flap, which is excised by ERCC1-XPF – a heterodimeric endonuclease that catalyzes a 5′ incision. A 9-nt gap is formed on the 5′ side of the lesion.
The RECQ1-PARP1 relationship regulates 5′ gap formation, suggesting that RECQ1 may be a PARP1 biological inhibitor. In addition, in the presence of PARP1 (independent of its poly ADP-ribosylation activity), RECQ1 significantly increased its binding to the substrate. Therefore, RECQ1 requires PARP1 to initiate this sub-pathway but inhibits PARP1’s activation.
Once the 9-nt gap is formed, POL δ/ε adds the appropriate nucleotides. PCNA, FEN1, and RPA are needed for similar functions as the other LP-BER pathways. RPA may also stimulate RECQ1. Ligation may be completed using either LIG1 or LIG3; however, the ligase type has not been identified. From the initial damage site, twenty nucleotides are incorporated.
Although the pathway was first discovered using plasmid-based studies, experiments conducted using genomic DNA showed this pathway can be applied to repair initiated by either monofunctional or bifunctional glycosylases and validated RECQ1-PARP1 regulation. Possible advantages of this pathway would be allowing for repair in PNKP-deficient mutants or repair after 8-oxodG incorporation by POL β/λ in high oxidative stress conditions. Although POL δ/ε has 3′ to 5′ proofreading capabilities, it cannot correct 8-oxodG lesions since the bypass efficiency is 60–70% [28] and cannot correct the 3′-terminal 8-oxodG [24]. APE1 and TDP1 may remove these 3′ modifications but have only been tested in in vitro experiments [2].
1.3. Nucleotide Incision Repair (NIR)
In 2002, Ischenko and Saparbaev discovered a glycosylase-independent repair pathway, which they named nucleotide incision repair (Figure 1f) [29]. APE1 creates an incision at the damage site by hydrolyzing the phosphodiester bond. This results in a damaged deoxynucleotide with 5′-PO4 and 3′-OH groups [30]. FEN1 and PCNA cleave the dangling nucleotide. Like SN-BER, POL β incorporates the new nucleotide, and LIG3/XRCC1 seals the nick. Advantages of this pathway include that it avoids the formation of toxic intermediates and explains DNA repair proficiency in glycosylase-deficient mutants [29]. However, it is crucial to consider that glycosylases may have a stronger binding affinity to the DNA substrate than APE1, making BER the preferred pathway. In addition, it is evolutionarily conserved, supporting the existence of a back-up repair pathway in organisms. Lastly, it may serve as a new physiological target for LP-BER but is currently not confirmed. It has been suggested that BER and NIR work together to provide genome stability since the rate of cleaving a dihydrouridine-containing (DHU) substrate by APE1 in NIR is comparable to the rate of cleaving an AP site in BER [30].
Although glycosylases (in BER) and APE1 (in NIR) both recognize damaged DNA, they have different structural DNA-binding sites and different amino acid residues that are involved in recognizing the damaged nucleotide [31]. APE1 substrate specificity factors the enzyme bending the damaged DNA, the damaged nucleotide turning inside out from the helix, and having specific contact between the base and the enzyme’s active site. In addition, GC-rich regions are preferred targets for BER, but do not all contain regions for Apn1-binding [32].
2. Transcription-Associated
DNA damage significantly threatens DNA replication and transcription if left unrepaired or repaired improperly. Since lesions placed on the non-transcribed strand have little or no effect on elongating RNA polymerase II (RNAPIIo—the hyper phosphorylated form of the polymerase) transcription, all cases discussed place the lesion on the transcribed strand. During transcription, RNAPIIo often encounters DNA lesions in the transcribed strand, causing RNAPIIo to pause or stall at the sites of the lesion; this could be the source of transcription-stress and mutagenesis, leading to varieties of disease phenotypes including aging and age-related pathologies. In normal cells, transcription-blocking lesions could be repaired by the conserved transcription-coupled repair (TCR) pathway that preferentially repairs DNA lesions from the transcribed strand of the active gene. UV irradiation or other genotoxic stressors block RNAPIIo. In mammalian cells, this signals TCR to repair bulky DNA lesions [33]. Several recent publications suggest the existence of a dedicated TCR pathway to repair oxidative DNA lesions [34][35][36].
2.1. Apurinic/Apyrimidinic (AP) Sites
AP sites are the most frequently generated lesions in the cell (approximately 10,000 per cell per day), predominantly caused by spontaneous hydrolysis of the glycosidic bond between deoxyribose and nucleobase or the damaged base removed by the DNA glycosylase (Figure 1) [37]. In vitro assays show that mammalian RNAPIIo assists AP site repair by blocking transcription on the transcribed strand [36].
2.2. Pyrimidine Modifications
Pyrimidines, such as cytosine modification products and their oxidized derivatives, are common epigenetic modifications that have a limited impact on transcription in eukaryotes. RNAPIIo is either bypassed or slightly paused during elongation and only slightly compromises transcription fidelity. Certain thymine modifications caused by components of cigarette smoke strongly impede transcription, suggesting that the position of modifications has an impact on transcription [38]. Uridine that arises in DNA because of cytosine deamination does not stall transcription, but RNAPIIo incorporates adenine opposite uracil during transcription. Template strand containing multiple uridines is transcribed but with low fidelity, possibly due to misincorporation. Two other oxidized pyrimidine lesions, 5′-OH cytosine and thymidine glycol, are bypassed by RNAPIIo with slight pause [39].
2.3. Purine Modifications
Purine oxidation generates a wide range of products; 8-oxoguanine (8-oxodG) is the most abundant, which can be further oxidized to non-bulky hydantoin lesions, such as guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp) [40]. RNAPIIo bypasses 8-oxodG lesions predominantly with misincorporation of adenine [36][41][42], but the oxidation products of 8-oxodG block RNAPIIo and favor misincorporation of purines [43][44]. Oxidation of a purine nucleotide by an OH radical can result in a crosslink between the nucleobase and deoxyribose, leading to the formation of CydA and CydG; both have high effects on transcription by causing RNAPIIo stalling [45]. Although uracil and cytosine are incorporated opposite CydA and CydG, respectively, adenine is incorporated in the following position, independent of the template. Consequently, this causes structural changes that make it difficult for RNAPIIo to recognize the 3′ end for continuous extension.
2.4. Single-Strand Breaks
Most DNA lesions in the cells (~75%) are SSBs arising from oxidative damage during metabolism or processing of AP sites. A nick in the template strand with damaged termini inhibits RNAP synthesis. If SSBs are not repaired rapidly and accurately, they can lead to cell death, chromosomal aberrations, and genetic mutations [46][47].
2.5. Evidence of BER-Linked Transcription in Mammalian Cells
Accurate repair of oxidative lesions is essential from the transcribing region to avoid the generation of mutant transcripts. However, most oxidized DNA bases only pause RNAPIIo elongation at the expense of transcriptional mutagenesis [48][49][50]. It is known that guanine is highly vulnerable to oxidation due to having a low redox potential, causing the formation of abundant 8-oxodG lesions, which only cause minor helix distortions, allowing RNAPIIo to bypass it unless processed by its specific glycosylase OGG1 [51].
NEIL2 is a particularly important glycosylase for the repair of damages linked to transcription because of its interaction with RNAPIIo, transcription–repair coupling factor CSB, and transcription factor IIH (TFIIH) [52][53]. In addition, it prefers lesions with the bubble structure that form during transcription. NEIL2 immunocomplex containing RNAPIIo and hnRNPU (RNA splicing factor) have been shown to repair oxidized base lesions from the transcribed strand [52]. Studies have shown that with age, DNA damage was accumulated in the actively transcribed genes in NEIL2-KD cells and oxidized base lesions in the transcribed genes in NEIL2 KO mice [54]. In addition, NEIL2 expression is independent of the cell cycle. Cumulatively, this suggests that NEIL2 is involved in BER linked to transcription.
NEIL2 is a bifunctional glycosylase that incises the associated AP sites through its AP lyase activity. During TET-TDG-mediated active demethylation, after TDG cleaves the N-glycosidic bond, NEIL2 serves only to process the AP site, suggesting that in certain contexts, NEIL2 preferentially processes AP sites and may substitute for APE1 [55]. A proposed model demonstrates that when RNAPIIo encounters AP sites or oxidative DNA lesions (Sp and Gh) during transcription, RNAPIIo stalls and recruits CSB and TFIIH to the repair sites. CSB brings NEIL2 through protein–protein interaction and stimulates NEIL2 activity, generating single-strand breaks with 3′-phosphate termini, which further stalls RNAPIIo. Retrograde movement or remodeling of the stalled RNAPIIo facilitates PNKP to replace the 3′-PO4 for the 3′-OH end; POL β then inserts the missing base, and LIG3-XRCC1 seals the nick and transcription is resumed (Figure 2).
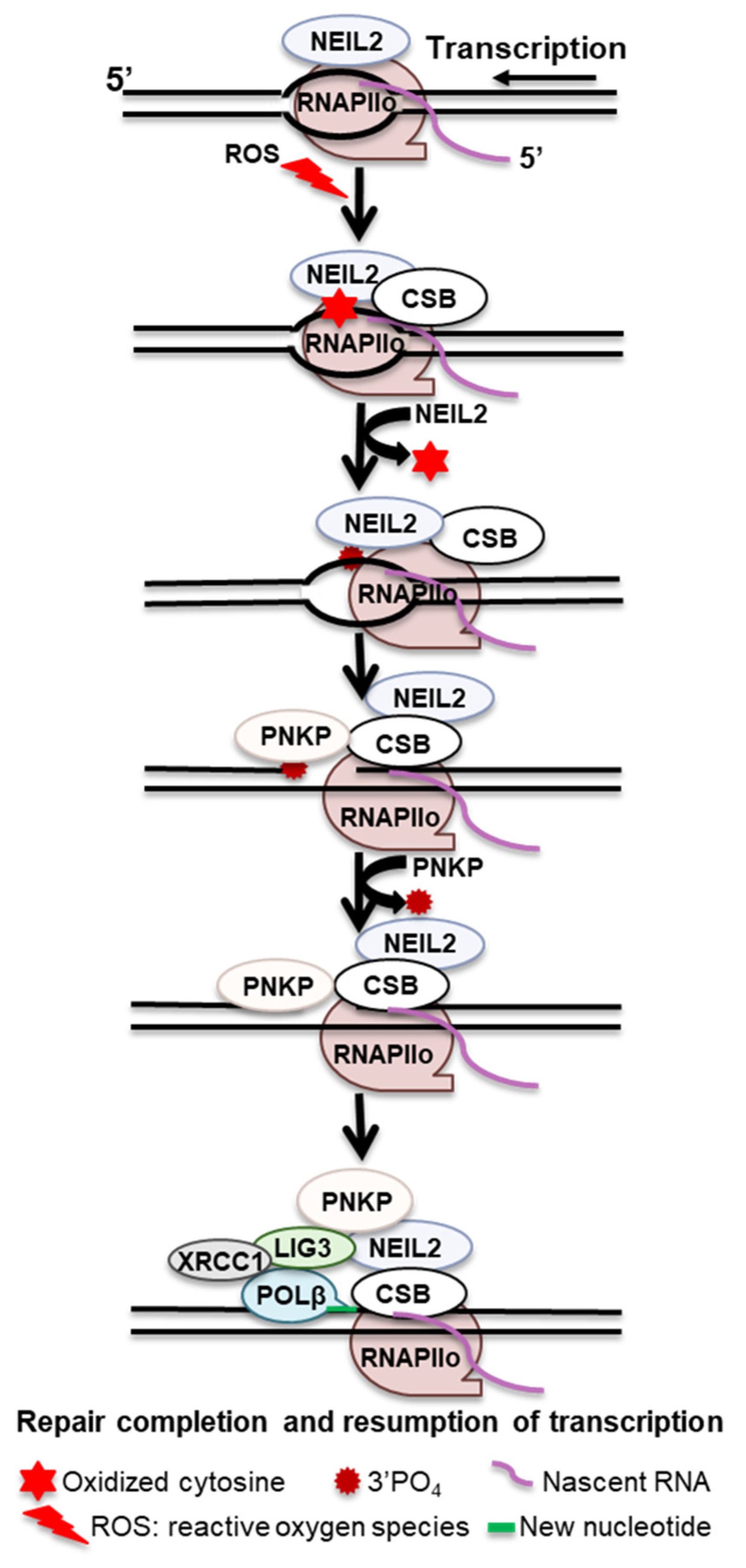
Figure 2. Proposed transcription-associated BER mechanism.
CSB and transcription-independent recruitment of OGG1 to 8-oxodG sites have been reported. However, recruiting CSB and XRCC1 to 8-oxodG sites depends on transcription, suggesting a transcription-dependent role of CSB beyond initial damage recognition [34]. Overall, CSB recognizes stalled RNAPIIo at OGG1-generated BER intermediates through its bifunctional glycosylase activity and recruits XRCC1 to mediate the coordination of subsequent repair factors for completion of SSB repair in transcribed genes.
3. Replication-Associated
Replication-associated repair refers to mechanisms that repair damaged DNA in coordination with the replisome. Some oxidized base lesions and abasic sites are potent blocks to replicative polymerases. The repair system does not always remove them before a DNA replication fork arrives. However, most oxidized lesions would not stall replicative polymerase but would cause mutations by inaccurate base pair during replication. Therefore, mutagenic base lesions must be repaired prior to replication to prevent mutation fixation. NEIL1 can repair oxidized lesions from structures mimicking the replication fork. In addition, its induced expression in the S phase of the cell cycle and stable physical and functional association with several proteins in the DNA replication complex cumulatively suggest that NEIL1 is part of the replication complex for repairing oxidized bases in the replicating template [56]. A model has been proposed that during replication, nonproductive NEIL1 binds to an oxidized lesion, such as 5-OHU or 5-OHC, in RPA-coated ssDNA; this stalls and regresses the fork that brings the lesion in the re-annealed duplex for faithful repair. The direct interaction of NEIL1 with RPA prevents DSB formation. NEIL1 repairs the lesion in association with replication machinery, including PCNA, RF-C, FEN1, POL δ, and LIG1; some are reported to stimulate NEIL1’s activity [57]. The functional association of NEIL1 with WRN or SMARCAL1 (both have ATPase activity) may coordinate fork regression and resolution to avoid fork collapse. Once repair is completed, replication resumes [58].
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263.
- Caglayan, M.; Horton, J.K.; Dai, D.P.; Stefanick, D.F.; Wilson, S.H. Oxidized nucleotide insertion by pol beta confounds ligation during base excision repair. Nat. Commun. 2017, 8, 14045.
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490.
- Parsons, J.L.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. DNA polymerase beta promotes recruitment of DNA ligase III alpha-XRCC1 to sites of base excision repair. Biochemistry 2005, 44, 10613–10619.
- Parsons, J.L.; Dianova, I.I.; Boswell, E.; Weinfeld, M.; Dianov, G.L. End-damage-specific proteins facilitate recruitment or stability of X-ray cross-complementing protein 1 at the sites of DNA single-strand break repair. FEBS J. 2005, 272, 5753–5763.
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schar, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670.
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996, 24, 4387–4394.
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117.
- Schermerhorn, K.M.; Delaney, S. A chemical and kinetic perspective on base excision repair of DNA. Acc. Chem. Res. 2014, 47, 1238–1246.
- Dalhus, B.; Laerdahl, J.K.; Backe, P.H.; Bjoras, M. DNA base repair--recognition and initiation of catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078.
- Margulies, C.M.; Samson, L.D. Alkyladenine DNA Glycosylases. In The Base Excision Repair Pathway, 1st ed.; Wilson, D.M., 3rd, Ed.; World Scientific: Hackensack, NJ, USA, 2017; pp. 189–218.
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair. 2019, 83, 102673.
- Quinones, J.L.; Demple, B. When DNA repair goes wrong: BER-generated DNA-protein crosslinks to oxidative lesions. DNA Repair. 2016, 44, 103–109.
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153.
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair. 2013, 12, 620–636.
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat. Res. 2010, 685, 80–89.
- Mortusewicz, O.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Differential recruitment of DNA Ligase I and III to DNA repair sites. Nucleic Acids Res. 2006, 34, 3523–3532.
- Liu, Y.; Beard, W.A.; Shock, D.D.; Prasad, R.; Hou, E.W.; Wilson, S.H. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J. Biol. Chem. 2005, 280, 3665–3674.
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, W.A.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell Biol. 2005, 25, 9350–9359.
- Sakurai, S.; Kitano, K.; Yamaguchi, H.; Hamada, K.; Okada, K.; Fukuda, K.; Uchida, M.; Ohtsuka, E.; Morioka, H.; Hakoshima, T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005, 24, 683–693.
- Chagovetz, A.M.; Sweasy, J.B.; Preston, B.D. Increased activity and fidelity of DNA polymerase beta on single-nucleotide gapped DNA. J. Biol. Chem. 1997, 272, 27501–27504.
- Podlutsky, A.J.; Dianova, I.I.; Podust, V.N.; Bohr, V.A.; Dianov, G.L. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001, 20, 1477–1482.
- Flood, C.L.; Rodriguez, G.P.; Bao, G.; Shockley, A.H.; Kow, Y.W.; Crouse, G.F. Replicative DNA polymerase delta but not epsilon proofreads errors in Cis and in Trans. PLoS Genet. 2015, 11, e1005049.
- DeMott, M.S.; Zigman, S.; Bambara, R.A. Replication protein A stimulates long patch DNA base excision repair. J. Biol. Chem. 1998, 273, 27492–27498.
- Pascucci, B.; Stucki, M.; Jónsson, Z.O.; Dogliotti, E.; Hübscher, U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J. Biol. Chem. 1999, 274, 33696–33702.
- Woodrick, J.; Gupta, S.; Camacho, S.; Parvathaneni, S.; Choudhury, S.; Cheema, A.; Bai, Y.; Khatkar, P.; Erkizan, H.V.; Sami, F.; et al. A new sub-pathway of long-patch base excision repair involving 5′ gap formation. EMBO J. 2017, 36, 1605–1622.
- Fazlieva, R.; Spittle, C.S.; Morrissey, D.; Hayashi, H.; Yan, H.; Matsumoto, Y. Proofreading exonuclease activity of human DNA polymerase delta and its effects on lesion-bypass DNA synthesis. Nucleic Acids Res. 2009, 37, 2854–2866.
- Ischenko, A.A.; Saparbaev, M.K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002, 415, 183–187.
- Timofeyeva, N.A.; Koval, V.V.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Kinetic mechanism of human apurinic/apyrimidinic endonuclease action in nucleotide incision repair. Biochemistry 2011, 76, 273–281.
- Kuznetsova, A.A.; Matveeva, A.G.; Milov, A.D.; Vorobjev, Y.N.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. Substrate specificity of human apurinic/apyrimidinic endonuclease APE1 in the nucleotide incision repair pathway. Nucleic Acids Res. 2018, 46, 11454–11465.
- Dyakonova, E.S.; Koval, V.V.; Lomzov, A.A.; Ishchenko, A.A.; Fedorova, O.S. Apurinic/apyrimidinic endonuclease Apn1 from Saccharomyces cerevisiae is recruited to the nucleotide incision repair pathway: Kinetic and structural features. Biochimie 2018, 152, 53–62.
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481.
- Menoni, H.; Wienholz, F.; Theil, A.F.; Janssens, R.C.; Lans, H.; Campalans, A.; Radicella, J.P.; Marteijn, J.A.; Vermeulen, W. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018, 46, 7747–7756.
- Chakraborty, A.; Tapryal, N.; Islam, A.; Mitra, S.; Hazra, T. Transcription coupled base excision repair in mammalian cells: So little is known and so much to uncover. DNA Repair. 2021, 107, 103204.
- Tornaletti, S.; Maeda, L.S.; Hanawalt, P.C. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem. Res. Toxicol. 2006, 19, 1215–1220.
- Liu, Z.J.; Martinez Cuesta, S.; van Delft, P.; Balasubramanian, S. Sequencing abasic sites in DNA at single-nucleotide resolution. Nat. Chem. 2019, 11, 629–637.
- Xu, L.; Wang, W.; Wu, J.; Shin, J.H.; Wang, P.; Unarta, I.C.; Chong, J.; Wang, Y.; Wang, D. Mechanism of DNA alkylation-induced transcriptional stalling, lesion bypass, and mutagenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E7082–E7091.
- Agapov, A.; Olina, A.; Kulbachinskiy, A. RNA polymerase pausing, stalling and bypass during transcription of damaged DNA: From molecular basis to functional consequences. Nucleic Acids Res. 2022, 50, 3018–3041.
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80.
- Kitsera, N.; Stathis, D.; Luhnsdorf, B.; Muller, H.; Carell, T.; Epe, B.; Khobta, A. 8-Oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1. Nucleic Acids Res. 2011, 39, 5926–5934.
- Pastoriza-Gallego, M.; Armier, J.; Sarasin, A. Transcription through 8-oxoguanine in DNA repair-proficient and Csb(-)/Ogg1(-) DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis 2007, 22, 343–351.
- Kolbanovskiy, M.; Chowdhury, M.A.; Nadkarni, A.; Broyde, S.; Geacintov, N.E.; Scicchitano, D.A.; Shafirovich, V. The Nonbulky DNA Lesions Spiroiminodihydantoin and 5-Guanidinohydantoin Significantly Block Human RNA Polymerase II Elongation in Vitro. Biochemistry 2017, 56, 3008–3018.
- Oh, J.; Fleming, A.M.; Xu, J.; Chong, J.; Burrows, C.J.; Wang, D. RNA polymerase II stalls on oxidative DNA damage via a torsion-latch mechanism involving lone pair-pi and CH-pi interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 9338–9348.
- Brooks, P.J.; Wise, D.S.; Berry, D.A.; Kosmoski, J.V.; Smerdon, M.J.; Somers, R.L.; Mackie, H.; Spoonde, A.Y.; Ackerman, E.J.; Coleman, K.; et al. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000, 275, 22355–22362.
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631.
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244.
- Charlet-Berguerand, N.; Feuerhahn, S.; Kong, S.E.; Ziserman, H.; Conaway, J.W.; Conaway, R.; Egly, J.M. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006, 25, 5481–5491.
- Kuraoka, I.; Suzuki, K.; Ito, S.; Hayashida, M.; Kwei, J.S.; Ikegami, T.; Handa, H.; Nakabeppu, Y.; Tanaka, K. RNA polymerase II bypasses 8-oxoguanine in the presence of transcription elongation factor TFIIS. DNA Repair. 2007, 6, 841–851.
- Saxowsky, T.T.; Meadows, K.L.; Klungland, A.; Doetsch, P.W. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18877–18882.
- Kuraoka, I.; Endou, M.; Yamaguchi, Y.; Wada, T.; Handa, H.; Tanaka, K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J. Biol. Chem. 2003, 278, 7294–7299.
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011, 286, 6006–6016.
- Aamann, M.D.; Hvitby, C.; Popuri, V.; Muftuoglu, M.; Lemminger, L.; Skeby, C.K.; Keijzers, G.; Ahn, B.; Bjoras, M.; Bohr, V.A.; et al. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 2014, 135, 1–14.
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648.
- Schomacher, L.; Han, D.; Musheev, M.U.; Arab, K.; Kienhofer, S.; von Seggern, A.; Niehrs, C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016, 23, 116–124.
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099.
- Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes 2017, 8, 175.
- Dutta, A.; Yang, C.; Sengupta, S.; Mitra, S.; Hegde, M.L. New paradigms in the repair of oxidative damage in human genome: Mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell Mol. Life Sci. 2015, 72, 1679–1698.
More
Information
Subjects:
Biochemistry & Molecular Biology; Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
26 Sep 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No