Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maryam Nakhjavani | -- | 1387 | 2023-09-18 15:25:05 | | | |
| 2 | Jessie Wu | + 5 word(s) | 1392 | 2023-09-19 02:59:42 | | | | |
| 3 | Jessie Wu | + 5 word(s) | 1397 | 2023-09-19 03:00:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Giles, B.; Nakhjavani, M.; Wiesa, A.; Knight, T.; Shigdar, S.; Samarasinghe, R.M. Immunotherapies Targeting the Tumour Microenvironment. Encyclopedia. Available online: https://encyclopedia.pub/entry/49343 (accessed on 05 July 2026).
Giles B, Nakhjavani M, Wiesa A, Knight T, Shigdar S, Samarasinghe RM. Immunotherapies Targeting the Tumour Microenvironment. Encyclopedia. Available at: https://encyclopedia.pub/entry/49343. Accessed July 05, 2026.
Giles, Breanna, Maryam Nakhjavani, Andrew Wiesa, Tareeque Knight, Sarah Shigdar, Rasika M. Samarasinghe. "Immunotherapies Targeting the Tumour Microenvironment" Encyclopedia, https://encyclopedia.pub/entry/49343 (accessed July 05, 2026).
Giles, B., Nakhjavani, M., Wiesa, A., Knight, T., Shigdar, S., & Samarasinghe, R.M. (2023, September 18). Immunotherapies Targeting the Tumour Microenvironment. In Encyclopedia. https://encyclopedia.pub/entry/49343
Giles, Breanna, et al. "Immunotherapies Targeting the Tumour Microenvironment." Encyclopedia. Web. 18 September, 2023.
Copy Citation
Brain tumour malignancies, especially glioblastoma multiform (GBM), characteristically reflect an abysmal disease prognosis with high mortality rates in both adult and paediatric populations. In adults, GBM is the most common and aggressive primary brain tumour, accounting for 15% of central nervous system (CNS) tumours, and 50.1% of malignancies, representing the most frequent form of primary malignant brain tumour.
glioblastoma
tumour microenvironment
aptamer
therapeutic
cancer
drug development
blood brain barrier
BBB
Immunotherapy
1. Introduction
The expanding knowledge of the glioblastoma multiform (GBM) tumour microenvironment (TME) has identified various potential therapeutic targets with a dominance of the development of several novel immunotherapies, including checkpoint inhibitors, mAbs, chimeric antigen receptor (CAR) modified T cells, and peptide or dendritic vaccines (Figure 1). While immunotherapies have proven efficacy in improving patient median overall survival (MOS) for other solid tumours, this is yet to be achieved for GBM. The challenge lies in trying to cross the blood-brain barrier (BBB) without inducing severe adverse events, addressing tumour heterogeneity, and overcoming the advanced immunosuppressive TME.
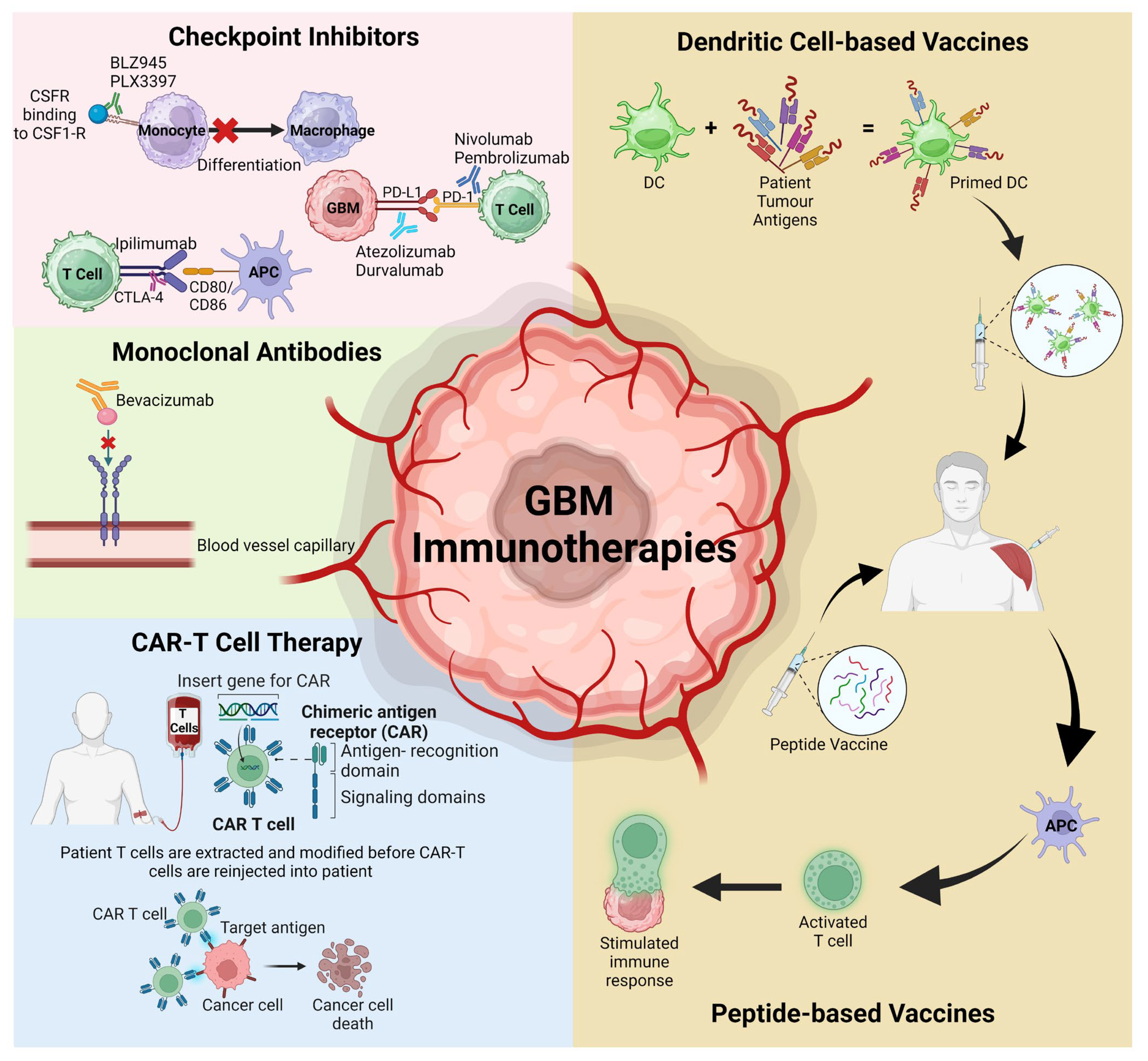
Figure 1. Schematic representation of the various immunotherapies discussed that have recently targeted the GBM TME and advanced to clinical trials. APC; antigen-presenting cells, CAR-T; chimeric antigen receptor T cells, CSFR; colony-stimulating factor receptor, DC; dendritic cells, PD-1; programmed cell death protein 1, PD-L1; programmed death ligand protein 1. Figure adapted from Kreatsoulas et al. and created using BioRender [1][2].
2. Checkpoint Inhibitors
Immune checkpoint inhibitors act to prevent co-inhibitory signals or mimic co-stimulatory signals as a response of controlling T cell functions upon major histocompatibility complex (MHC) class I/II antigens binding to T cell receptors. To date, the checkpoint blockade has shown minimal benefits in improving adult and paediatric GBM, either alone or when combined with the mAb bevacizumab [3]. The most common of these targets are colony-stimulating factor-1 receptor (CSF-1R), PD-L1 and cytotoxic T-lymphocyte associated protein-4 (CTLA-4) receptors found within GBM TME.
2.1. Colony Stimulating Factor-1 Receptor (CSF-1R)
Using a genetic mouse model, the CSF-1R inhibitor BLZ945 caused an acquired resistance via increased macrophage-derived insulin-like growth factor-1 (IGF-1) and tumour cell IGF-1 receptors, ultimately enhancing the PI3K pathway. In combination with IGF-1R or PI3K blockers, an improved OS was detected compared to monotherapy; however, greater than 50% of mice experienced GBM recurrence [4]. In a GBM orthotopic immunocompetent mouse model, BLZ945 monotherapy did not improve median OS, but in combination with RT, a significantly improved overall survival (OS) was detected [5]. The other CSF-1R inhibitor PLX3397 combined with RT decreased tumour size by 100-fold and improved median survival compared to RT alone. PLX3397 enhanced the efficacy of RT by preventing recruited monocytes from differentiating into the immunosuppressive macrophages [6]. In a phase II clinical trial (NCT01349036), recurrent GBM patients receiving PLX3397 (1000 mg/day), showed no therapeutic effect [7]. Monotherapy with CSF-1R inhibitors appears to be insufficient in overcoming the highly immunosuppressive microenvironment of GBM.
2.2. Programmed Cell Death Protein-1 (PD1) and Its Ligand Programmed Death Ligand Protein 1
PD-L1 is expressed in 88% of GMB tumours within the TME on microglia and tumour-associated macrophages (TAMs) [8]. High PD-L1 expression on neurons in adjacent brain tissue and low within GBM cells reflects better patient outcomes. Increased PD-L1 expression within glioma cells contributes to a higher tumour grade and worse patient outcomes [9]. Given the immunosuppressive nature of GBM, PD-1 inhibitors including nivolumab and pembrolizumab have been unsuccessful within clinical settings, despite showing results in other cancers. PD-L1 inhibitors atezolizumab or durvolumab have been successful in GBM cases with specific DNA-repair mismatch defects or biallelic mismatch repair deficiencies [10][11]. The Keynote-028 trial assessed pembrolizumab monotherapy in 26 recurrent GBM patients, but minimal survival benefits were reported, with a median progression-free survival (PFS) and OS of 2.8 and 14.4 months, respectively [12]. A combination of nivolumab and bevacizumab showed no significant improvement in a phase II clinical trial in recurrent GBM (NCT03452579) [13]. However, neoadjuvant pembrolizumab with adjuvant therapy post-surgery significantly improved OS in recurrent GBM compared to adjuvant therapy, post-surgical pembrolizumab (13.2 vs. 6.3 months). This neoadjuvant therapy enhanced local and systemic immune responses in patients with an enhanced T cell clonal expansion and a decreased peripheral blood T cell PD-1 expression [14]. Additional combined approaches will be required to improve the efficacy of PD-1 and PD-L1 checkpoint inhibitors.
2.3. Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4)
The overexpression of CTLA-4 contributes to worse patient outcomes in higher-grade brain tumours, including GBM [15]. One experimental arm of the CheckMate 143 clinical trial involved a combination of 3 mg/kg nivolumab with either 1 or 3 mg/kg ipilimumab, a checkpoint inhibitor targeting CTLA-4 [16]. However, nivolumab alone was more tolerable and efficacious than the combined approach, with a greater percentage of patients discontinuing from adverse events, including fatigue and diarrhoea [16].
3. Chimeric Antigen Receptor T (CAR-T) Cell Therapy
Since the generation of the first CAR-T cells in 1987, remarkable therapeutic efficacy has been shown in haematological cancers and more recently in some solid tumours [17][18]. The challenging concept in achieving therapeutic efficacy in GBM is overcoming the immunosuppressive TME, surface tumour antigen heterogeneity, and difficulty in trafficking CAR-T cells from a patient’s blood to tumour sites [19]. Genetically engineered CAR-T cells are artificial fusion proteins that specifically bind to tumour antigens and overcome defective neoantigen presentation, lack of immune priming and low tumour mutational load, which hinders immune inhibition in GBM to induce an anti-tumour T-cell [20].
In a first-in-human study of single-dose intravenous delivery of epidermal growth factor receptor-variant III (EGFRvIII) engineered CAR-T-EGFRvIII cells to 10 recurrent GBM patients, initial reports revealed a safe infusion with no off-tumour toxicity or cytokine release syndrome. However, pathological in situ analysis of GBM TME revealed the activation of an adaptive immunosuppressive response by an enhanced infiltration of Tregs and expression of inhibitory molecules interleukin-10, PD-L1, and transforming growth factor-beta (TGF-β) [21].
Another first-in-human trial reported three recurrent GBM patients treated through 12 local infusions with CAR-T cells, autologous CD8+ CTLs targeting interleukin 13 receptor subunit alpha 2 (IL13Rα2). This treatment was well tolerated with only temporary central nervous system (CNS) inflammation reported, despite only two patients exhibiting anti-tumour responses, either through reduced IL13Rα2 expression or an increased necrotic tumour volume at the delivery site [22].
Upon IL13Rα2-targeted CAR-T cells administered to IDH-wild type, unmethylated MGMT gene expressed recurrent GBM patients, all intracranial and spinal tumours regressed with enhanced levels of immune cells detected in the cerebrospinal fluid, demonstrating immune system activation for up to 7.5 months. Despite an initial complete response and prevention of recurrence, treatment failed to control tumour progression within distant sites [23].
4. Vaccinations
Several GBM-targeting vaccine candidates are in the early stages of development. DC-based vaccines use collected autologous DCs, prime them ex vivo with patient tumour antigens and are administered intradermally. Alternatively, peptide-based vaccines are tumour-specific antigens trafficked into patients for antigen-presenting cells to present to T cells and stimulate an immune response [24].
Forty-one recurrent GBM adult patients post-surgery received a heat-shock protein peptide complex-96 vaccine, and 90.2% of patients reached the primary endpoint of 6 months, while 29.3% survived greater than 12 months [25]. However, 66% of patients were lympho-penic prior to treatment which significantly impacted the OS. Vaccine toxicity was minimal, and no treatment-related deaths were reported [25].
Autologous DC vaccines loaded with tumour lysates isolated from resected GBM tumours in 56 adult and paediatric patients demonstrated safety and induced long-term survival in patients, despite only a marginal improvement of median PFS and OS rates measured from prior to second surgery, at 3 and 9.6 months, respectively [26]. While immune responses were not assessed, paediatrics and adults under 35 years of age demonstrated a better OS than older adults; similarly, patients who experienced a greater extent of tumour resection had better PFS and OS rates. Only mild adverse events were reported, although one patient experienced vaccine-induced grade IV neurotoxicity given the large residual tumour size, while another developed GBM metastasis within spine and lungs [26].
5. Monoclonal Antibodies (mAbs)
mAbs, due to their high specificity and sensitivity to biological targets, have been widely used to treat various cancers in order to elicit immunotherapeutic and anti-angiogenic responses in GBM against growth factor receptors EGFR and VEGFR [27]. Bevacizumab, the anti-vascular endothelial growth factor (VEGF) mAb, inhibits angiogenesis, metastases, DC maturation, antigen presentation and lymphocyte trafficking into tumours [24]. Bevacizumab is currently the only Food and Drug Administration (FDA) approved mAb for GBM treatment, but like most antibodies, due to its large size, is unable to cross the BBB. A systematic analysis has revealed that bevacizumab can prolong OS of recurrent GBM patients by approximately 4 months post standard-of-care therapies, but not for primary GBM. Seventy-four percent of the patients experienced grade 3 or higher toxicity, including hypertension, lymphopenia, leukopenia, neutropenia, or thromboembolic events [28].
References
- BioRender. Available online: https://www.biorender.com/ (accessed on 20 August 2023).
- Kreatsoulas, D.; Bolyard, C.; Wu, B.X.; Cam, H.; Giglio, P.; Li, Z. Translational landscape of glioblastoma immunotherapy for physicians: Guiding clinical practice with basic scientific evidence. J. Hematol. Oncol. 2022, 15, 80.
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142.
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018.
- Almahariq, M.F.; Quinn, T.J.; Kesarwani, P.; Kant, S.; Miller, C.R.; Chinnaiyan, P. Inhibition of Colony-Stimulating Factor-1 Receptor Enhances the Efficacy of Radiotherapy and Reduces Immune Suppression in Glioblastoma. Vivo 2021, 35, 119–129.
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. J. Neuro-Oncol. 2016, 18, 797–806.
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. J. Neuro-Oncol. 2016, 18, 557–564.
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. J. Neuro-Oncol. 2015, 17, 1064–1075.
- Liu, Y.; Carlsson, R.; Ambjørn, M.; Hasan, M.; Badn, W.; Darabi, A.; Siesjö, P.; Issazadeh-Navikas, S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J. Neurosci. 2013, 33, 14231–14245.
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Clin. Oncol. 2016, 34, 2206–2211.
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016, 6, 1230–1236.
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327.
- Ahluwalia, M.S.; Rauf, Y.; Li, H.; Wen, P.Y.; Peereboom, D.M.; Reardon, D.A. Randomized phase 2 study of nivolumab (nivo) plus either standard or reduced dose bevacizumab (bev) in recurrent glioblastoma (rGBM). Clin. Oncol. 2021, 39, 2015.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486.
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020, 20, 7.
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. J. Neuro-Oncol. 2018, 20, 674–686.
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968.
- Styczyński, J. A brief history of CAR-T cells: From laboratory to the bedside. Acta Haematol. Pol. 2020, 51, 2–5.
- Wang, X.; Lu, J.; Guo, G.; Yu, J. Immunotherapy for recurrent glioblastoma: Practical insights and challenging prospects. Cell Death Dis. 2021, 12, 299.
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. J. Neuro-Oncol. 2018, 20, 1429–1438.
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984.
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569.
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181.
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. J. Neuro-Oncol. 2014, 16, 274–279.
- De Vleeschouwer, S.; Fieuws, S.; Rutkowski, S.; Van Calenbergh, F.; Van Loon, J.; Goffin, J.; Sciot, R.; Wilms, G.; Demaerel, P.; Warmuth-Metz, M.; et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 3098–3104.
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532.
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neuro-Oncol. 2017, 133, 455–467.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
607
Revisions:
3 times
(View History)
Update Date:
19 Sep 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No