 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Satoru Matsuda | -- | 3396 | 2023-09-07 15:49:38 | | | |
| 2 | Peter Tang | Meta information modification | 3396 | 2023-09-08 04:42:19 | | |
Video Upload Options
A brain-enriched multi-domain scaffolding protein, neurobeachin has been identified as a candidate gene for autism patients. Mutations in the synaptic adhesion protein cell adhesion molecule 1 (CADM1) are also associated with autism spectrum disorder, a neurodevelopmental disorder of uncertain molecular origin. Potential roles of neurobeachin and CADM1 have been suggested to a function of vesicle transport in endosomal trafficking. It seems that protein kinase B (AKT) and cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) have key roles in the neuron membrane trafficking involved in the pathogenesis of autism. Attention deficit hyperactivity disorder (ADHD) is documented to dopaminergic insufficiencies, which is attributed to synaptic dysfunction of dopamine transporter (DAT). AKT is also essential for the DAT cell-surface redistribution.
1. Introduction
2. Relationship between Autisms and Neurobeachin
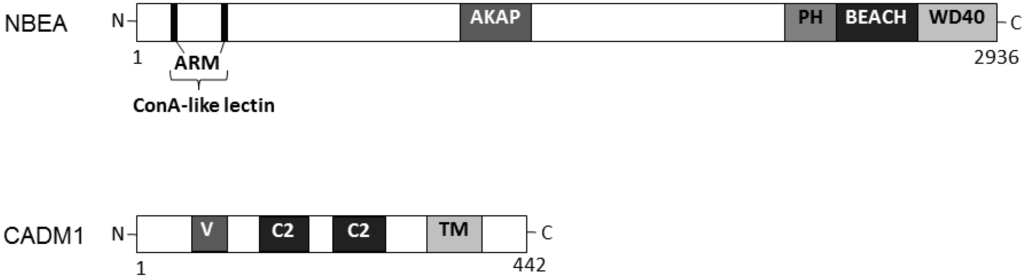
3. Relationship between Autisms and Cell Adhesion Molecule 1 (CADM1)
4. Relationship between Attention Deficit/Hyperactivity Disorder (ADHD) and Dopamine Transporter (DAT)
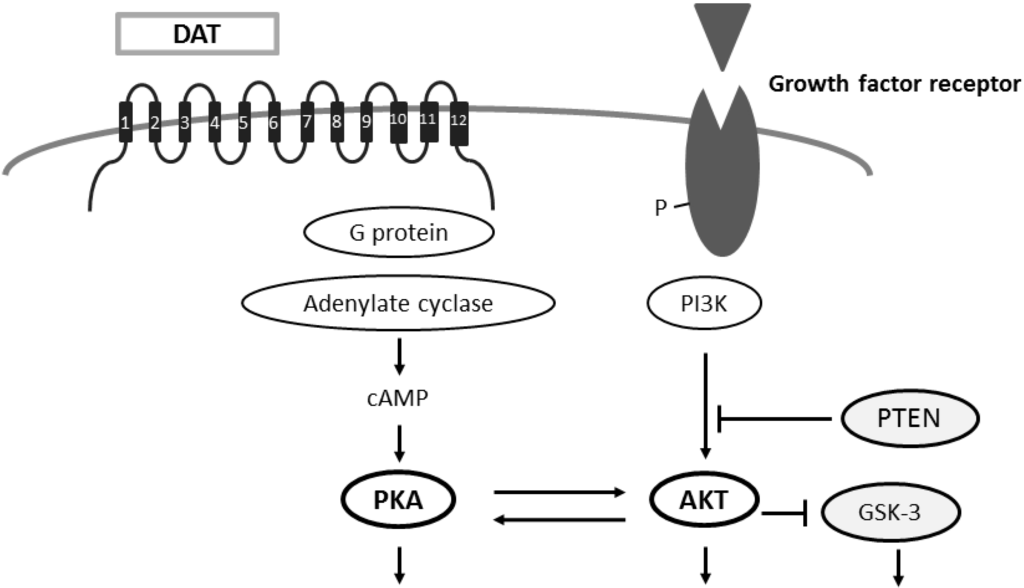
5. Neuronal Membrane Trafficking Involved in Autisms and ADHD Regulated by Several Protein Kinases
6. Interplay of the Kinases Involved in Autisms and ADHD
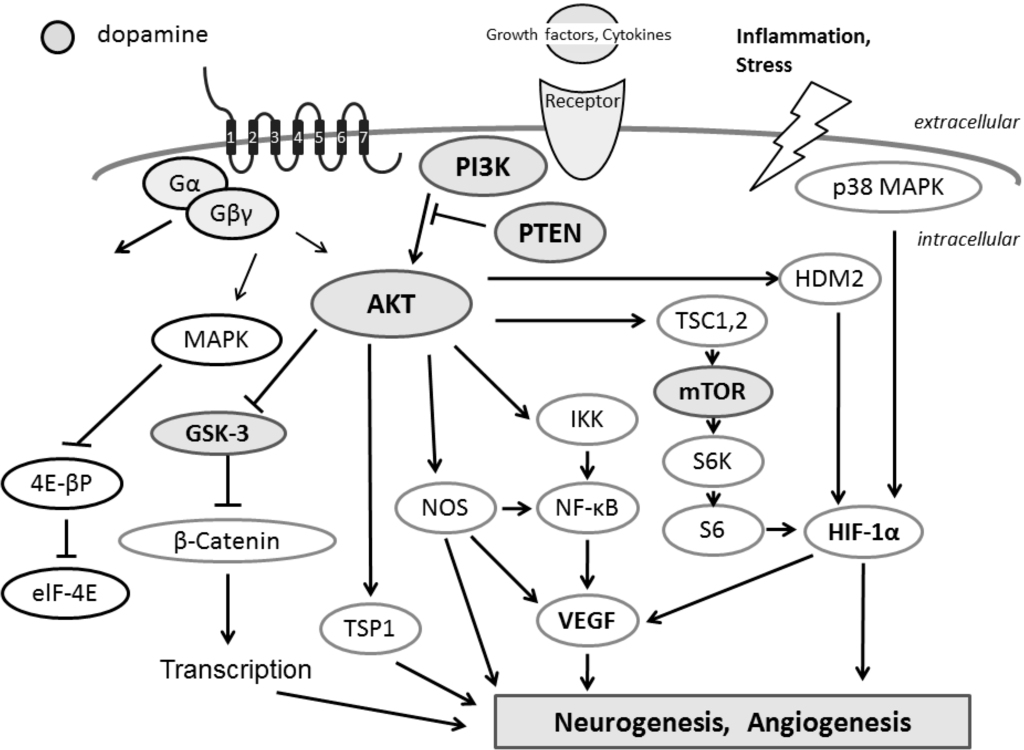
7. Diets May Contribute to the Improved Membrane Trafficking in Autisms and ADHD via the Modulation of AKT and PKA Signaling
References
- Huang, C.; Rajfur, Z.; Yousefi, N.; Chen, Z.; Jacobson, K.; Ginsberg, M.H. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 2009, 11, 624–630.
- Bishop, N.E. Dynamics of endosomal sorting. Int. Rev. Cytol. 2003, 232, 1–57.
- Mizutani, R.; Yamauchi, J.; Kusakawa, S.; Nakamura, K.; Sanbe, A.; Torii, T.; Miyamoto, Y.; Tanoue, A. Sorting nexin 3, a protein up-regulated by lithium, contains a novel phosphatidylinositol-binding sequence and mediates neurite outgrowth in N1E-115 cells. Cell Signal. 2009, 21, 1586–1594.
- Koutelou, E.; Sato, S.; Tomomori-Sato, C.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Kokkinaki, M.; Conaway, R.C.; Conaway, J.W.; Moschonas, N.K. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J. Biol. Chem. 2008, 283, 3846–3853.
- Kanamarlapudi, V. Centaurin-α1 and KIF13B kinesin motor protein interaction in ARF6 signalling. Biochem. Soc. Trans. 2005, 33, 1279–1281.
- Venkateswarlu, K.; Gunn-Moore, F.; Tavaré, J.M.; Cullen, P.J. EGF-and NGF-stimulated translocation of cytohesin-1 to the plasma membrane of PC12 cells requires PI 3-kinase activation and a functional cytohesin-1 PH domain. J. Cell Sci. 1999, 112, 1957–1965.
- Woo, J.; Chae, Y.K.; Jang, S.J.; Kim, M.S.; Baek, J.H.; Park, J.C.; Trink, B.; Ratovitski, E.; Lee, T.; Park, B.; et al. Membrane trafficking of AQP5 and cAMP dependent phosphorylation in bronchial epithelium. Biochem. Biophys. Res. Commun. 2008, 366, 321–327.
- Wojtal, K.A.; Hoekstra, D.; van Ijzendoorn, S.C. cAMP-dependent protein kinase A and the dynamics of epithelial cell surface domains: Moving membranes to keep in shape. Bioessays 2008, 30, 146–155.
- Foletti, D.L.; Prekeris, R.; Scheller, R.H. Generation and maintenance of neuronal polarity: Mechanisms of transport and targeting. Neuron 1999, 23, 641–644.
- Horton, A.C.; Ehlers, M.D. Neuronal polarity and trafficking. Neuron 2003, 40, 277–295.
- Tang, D.; Wang, Y. Cell cycle regulation of Golgi membrane dynamics. Trends Cell Biol. 2013, 23, 296–304.
- Lecuit, T. Regulation of membrane dynamics in developing epithelia. Curr. Opin. Genet. Dev. 2003, 13, 351–357.
- Bowton, E.; Saunders, C.; Reddy, I.A.; Campbell, N.G.; Hamilton, P.J.; Henry, L.K.; Coon, H.; Sakrikar, D.; Veenstra-VanderWeele, J.M.; Blakely, R.D.; et al. SLC6A3 coding variant Ala559Val found in two autism probands alters dopamine transporter function and trafficking. Transl. Psychiatry 2014, 4, e464.
- Sakrikar, D.; Mazei-Robison, M.S.; Mergy, M.A.; Richtand, N.W.; Han, Q.; Hamilton, P.J.; Bowton, E.; Galli, A.; Veenstra-Vanderweele, J.; Gill, M.; et al. Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J. Neurosci. 2012, 32, 5385–5397.
- Leitner, Y. The co-occurrence of autism and attention deficit hyperactivity disorder in children—What do we know? Front. Hum. Neurosci. 2014, 8, 268.
- Kondapalli, K.C.; Prasad, H.; Rao, R. An inside job: How endosomal Na+/H+ exchangers link to autism and neurological disease. Front. Cell Neurosci. 2014, 8, 172.
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770.
- Zuko, A.; Kleijer, K.T.; Oguro-Ando, A.; Kas, M.J.; van Daalen, E.; van der Zwaag, B.; Burbach, J.P. Contactins in the neurobiology of autism. Eur. J. Pharmacol. 2013, 719, 63–74.
- Gu, W.; Lupski, J.R. CNV and nervous system diseases—What’s new? Cytogenet. Genome Res. 2008, 123, 54–64.
- Volders, K.; Nuytens, K.; Creemers, J.W. The autism candidate gene Neurobeachin encodes a scaffolding protein implicated in membrane trafficking and signaling. Curr. Mol. Med. 2011, 11, 204–217.
- Olszewski, P.K.; Rozman, J.; Jacobsson, J.A.; Rathkolb, B.; Strömberg, S.; Hans, W.; Klockars, A.; Alsiö, J.; Risérus, U.; Becker, L.; et al. Neurobeachin, a regulator of synaptic protein targeting, is associated with body fat mass and feeding behavior in mice and body-mass index in humans. PLoS Genet. 2012, 8, e1002568.
- Wang, X.; Herberg, F.W.; Laue, M.M.; Wullner, C.; Hu, B.; Petrasch-Parwez, E.; Kilimann, M.W. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J. Neurosci. 2000, 20, 8551–8565.
- Savelyeva, L.; Sagulenko, E.; Schmitt, J.G.; Schwab, M. The neurobeachin gene spans the common fragile site FRA13A. Hum. Genet. 2006, 118, 551–558.
- De Lozanne, A. The role of BEACH proteins in Dictyostelium. Traffic 2003, 4, 6–12.
- Vaccari, T.; Bilder, D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell 2005, 9, 687–698.
- Nuytens, K.; Tuand, K.; di Michele, M.; Boonen, K.; Waelkens, E.; Freson, K.; Creemers, J.W. Platelets of mice heterozygous for neurobeachin, a candidate gene for autism spectrum disorder, display protein changes related to aberrant protein kinase A activity. Mol. Autism 2013, 4, 43.
- Shamloula, H.K.; Mbogho, M.P.; Pimentel, A.C.; Chrzanowska-Lightowlers, Z.M.; Hyatt, V.; Okano, H.; Venkatesh, T.R. rugose (rg), a Drosophila A kinase anchor protein, is required for retinal pattern formation and interacts genetically with multiple signaling pathways. Genetics 2002, 161, 693–710.
- Wang, K.; Hackett, J.T.; Cox, M.E.; van Hoek, M.; Lindstrom, J.M.; Parsons, S.J. Regulation of the neuronal nicotinic acetylcholine receptor by SRC family tyrosine kinases. J. Biol. Chem. 2004, 279, 8779–8786.
- Miller, A.C.; Voelker, L.H.; Shah, A.N.; Moens, C.B. Neurobeachin is required postsynaptically for electrical and chemical synapse formation. Curr. Biol. 2015, 25, 16–28.
- Chen, C.K.; Bregere, C.; Paluch, J.; Lu, J.F.; Dickman, D.K.; Chang, K.T. Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun. 2014, 5, 4246.
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337.
- Ribic, A.; Liu, X.; Crair, M.C.; Biederer, T. Structural organization and function of mouse photoreceptor ribbon synapses involve the immunoglobulin protein synaptic cell adhesion molecule 1. J. Comp. Neurol. 2014, 522, 900–920.
- Masuda, M.; Yageta, M.; Fukuhara, H.; Kuramochi, M.; Maruyama, T.; Nomoto, A.; Murakami, Y. The tumor suppressor protein TSLC1 is involved in cell-cell adhesion. J. Biol. Chem. 2002, 277, 31014–31019.
- Fujita, E.; Tanabe, Y.; Imhof, B.A.; Momoi, M.Y.; Momoi, T. A complex of synaptic adhesion molecule CADM1, a molecule related to autism spectrum disorder, with MUPP1 in the cerebellum. J. Neurochem. 2012, 123, 886–894.
- Fujita, E.; Dai, H.; Tanabe, Y.; Zhiling, Y.; Yamagata, T.; Miyakawa, T.; Tanokura, M.; Momoi, M.Y.; Momoi, T. Autism spectrum disorder is related to endoplasmic reticulum stress induced by mutations in the synaptic cell adhesion molecule, CADM1. Cell Death Dis. 2010, 1, e47.
- Patiño-Lopez, G.; Hevezi, P.; Lee, J.; Willhite, D.; Verge, G.M.; Lechner, S.M.; Ortiz-Navarrete, V.; Zlotnik, A. Human class-I restricted T cell associated molecule is highly expressed in the cerebellum and is a marker for activated NKT and CD8+ T lymphocytes. J. Neuroimmunol. 2006, 171, 145–155.
- Tanabe, Y.; Fujita, E.; Hayashi, Y.K.; Zhu, X.; Lubbert, H.; Mezaki, Y.; Senoo, H.; Momoi, T. Synaptic adhesion molecules in Cadm family at the neuromuscular junction. Cell Biol. Int. 2013, 37, 731–736.
- Murphy, C.M.; Christakou, A.; Daly, E.M.; Ecker, C.; Giampietro, V.; Brammer, M.; Smith, A.B.; Johnston, P.; Robertson, D.M.; MRC AIMS Consortium; et al. Abnormal functional activation and maturation of fronto-striato-temporal and cerebellar regions during sustained attention in autism spectrum disorder. Am. J. Psychiatry 2014, 171, 1107–1116.
- Zhiling, Y.; Fujita, E.; Tanabe, Y.; Yamagata, T.; Momoi, T.; Momoi, M.Y. Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem. Biophys. Res. Commun. 2008, 377, 926–929.
- Momoi, T.; Fujita, E.; Senoo, H.; Momoi, M. Genetic factors and epigenetic factors for autism: Endoplasmic reticulum stress and impaired synaptic function. Cell Biol. Int. 2009, 34, 13–19.
- Williams, Y.N.; Masuda, M.; Sakurai-Yageta, M.; Maruyama, T.; Shibuya, M.; Murakami, Y. Cell adhesion and prostate tumor-suppressor activity of TSLL2/IGSF4C, an immunoglobulin superfamily molecule homologous to TSLC1/IGSF4. Oncogene 2006, 25, 1446–1453.
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene × Environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front. Psychiatry 2014, 5, 53.
- Hauser, T.U.; Iannaccone, R.; Ball, J.; Mathys, C.; Brandeis, D.; Walitza, S.; Brem, S. Role of the medial prefrontal cortex in impaired decision making in juvenile attention-deficit/hyperactivity disorder. JAMA Psychiatry 2014, 71, 1165–1173.
- Pearlman, D.M.; Vora, H.S.; Marquis, B.G.; Najjar, S.; Dudley, L.A. Anti-basal ganglia antibodies in primary obsessive-compulsive disorder: Systematic review and meta-analysis. Br. J. Psychiatry 2014, 205, 8–16.
- Kirley, A.; Hawi, Z.; Daly, G.; McCarron, M.; Mullins, C.; Millar, N.; Waldman, I.; Fitzgerald, M.; Gill, M. Dopaminergic system genes in ADHD: Toward a biological hypothesis. Neuropsychopharmacology 2002, 27, 607–619.
- Grant, P.; Kuepper, Y.; Wielpuetz, C.; Hennig, J. Differential associations of dopamine-related polymorphisms with discrete components of reaction time variability: Relevance for attention deficit/hyperactivity disorder. Neuropsychobiology 2014, 69, 220–226.
- Somkuwar, S.S.; Darna, M.; Kantak, K.M.; Dwoskin, L.P. Adolescence methylphenidate treatment in a rodent model of attention deficit/hyperactivity disorder: Dopamine transporter function and cellular distribution in adulthood. Biochem. Pharmacol. 2013, 86, 309–316.
- Patel, J.; Mooslehner, K.A.; Chan, P.M.; Emson, P.C.; Stamford, J.A. Presynaptic control of striatal dopamine neurotransmission in adult vesicular monoamine transporter 2 (VMAT2) mutant mice. J. Neurochem. 2003, 85, 898–910.
- Kim, P.; Choi, C.S.; Park, J.H.; Joo, S.H.; Kim, S.Y.; Ko, H.M.; Kim, K.C.; Jeon, S.J.; Park, S.H.; Han, S.H.; et al. Chronic exposure to ethanol of male mice before mating produces attention deficit hyperactivity disorder-like phenotype along with epigenetic dysregulation of dopamine transporter expression in mouse offspring. J. Neurosci. Res. 2014, 92, 658–670.
- Hall, F.S.; Itokawa, K.; Schmitt, A.; Moessner, R.; Sora, I.; Lesch, K.P.; Uhl, G.R. Decreased vesicular monoamine transporter 2 (VMAT2) and dopamine transporter (DAT) function in knockout mice affects aging of dopaminergic systems. Neuropharmacology 2014, 76, 146–155.
- Greenwood, T.A.; Joo, E.J.; Shekhtman, T.; Sadovnick, A.D.; Remick, R.A.; Keck, P.E.; McElroy, S.L.; Kelsoe, J.R. Association of dopamine transporter gene variants with childhood ADHD features in bipolar disorder. Am. J. Med. Genet. B 2013, 162B, 137–145.
- Fosi, T.; Lax-Pericall, M.T.; Scott, R.C.; Neville, B.G.; Aylett, S.E. Methylphenidate treatment of attention deficit hyperactivity disorder in young people with learning disability and difficult-to-treat epilepsy: Evidence of clinical benefit. Epilepsia 2013, 54, 2071–2081.
- Meneses, A.; Perez-Garcia, G.; Ponce-Lopez, T.; Tellez, R.; Gallegos-Cari, A.; Castillo, C. Spontaneously hypertensive rat (SHR) as an animal model for ADHD: A short overview. Rev. Neurosci. 2011, 22, 365–371.
- Barth, V.; Need, A.B.; Tzavara, E.T.; Giros, B.; Overshiner, C.; Gleason, S.D.; Wade, M.; Johansson, A.M.; Perry, K.; Nomikos, G.G.; et al. In vivo occupancy of dopamine D3 receptors by antagonists produces neurochemical and behavioral effects of potential relevance to attention-deficit-hyperactivity disorder. J. Pharmacol. Exp. Ther. 2013, 344, 501–510.
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Logan, J.; Franceschi, D.; Maynard, L.; Ding, Y.S.; Gatley, S.J.; Gifford, A.; Zhu, W.; et al. Relationship between blockade of dopamine transporters by oral methylphenidate and the increases in extracellular dopamine: Therapeutic implications. Synapse 2002, 43, 181–187.
- Torres, G.E. The dopamine transporter proteome. J. Neurochem. 2006, 97, 3–10.
- Robbins, T.W. Dopamine and cognition. Curr. Opin. Neurol. 2003, 16, S1–S2.
- Nuytens, K.; Gantois, I.; Stijnen, P.; Iscru, E.; Laeremans, A.; Serneels, L.; van Eylen, L.; Liebhaber, S.A.; Devriendt, K.; Balschun, D.; et al. Haploinsufficiency of the autism candidate gene Neurobeachin induces autism-like behaviors and affects cellular and molecular processes of synaptic plasticity in mice. Neurobiol. Dis. 2013, 51, 144–151.
- Su, Y.; Balice-Gordon, R.J.; Hess, D.M.; Landsman, D.S.; Minarcik, J.; Golden, J.; Hurwitz, I.; Liebhaber, S.A.; Cooke, N.E. Neurobeachin is essential for neuromuscular synaptic transmission. J. Neurosci. 2004, 24, 3627–3636.
- Ohashi, M.; Huttner, W.B. An elevation of cytosolic protein phosphorylation modulates trimeric G-protein regulation of secretory vesicle formation from the trans-Golgi network. J. Biol. Chem. 1994, 269, 24897–24905.
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–221.
- Dell'Acqua, M.L.; Smith, K.E.; Gorski, J.A.; Horne, E.A.; Gibson, E.S.; Gomez, L.L. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur. J. Cell Biol. 2006, 85, 627–633.
- Röder, I.V.; Choi, K.R.; Reischl, M.; Petersen, Y.; Diefenbacher, M.E.; Zaccolo, M.; Pozzan, T.; Rudolf, R. Myosin Va cooperates with PKA RIα to mediate maintenance of the endplate in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 2031–2036.
- Niesmann, K.; Breuer, D.; Brockhaus, J.; Born, G.; Wolff, I.; Reissner, C.; Kilimann, M.W.; Rohlmann, A.; Missler, M. Dendritic spine formation and synaptic function require neurobeachin. Nat. Commun. 2011, 2, 557.
- Murakami, S.; Sakurai-Yageta, M.; Maruyama, T.; Murakami, Y. Trans-homophilic interaction of CADM1 activates PI3K by forming a complex with MAGuK-family proteins MPP3 and Dlg. PLoS One 2014, 9, e82894.
- Masuda, M.; Maruyama, T.; Ohta, T.; Ito, A.; Hayashi, T.; Tsukasaki, K.; Kamihira, S.; Yamaoka, S.; Hoshino, H.; Yoshida, T.; et al. CADM1 interacts with Tiam1 and promotes invasive phenotype of human T-cell leukemia virus type I-transformed cells and adult T-cell leukemia cells. J. Biol. Chem. 2010, 285, 15511–15522.
- Montgomery, J.M.; Zamorano, P.L.; Garner, C.C. AGUKs in synapse assembly and function: An emerging view. Cell Mol. Life Sci. 2004, 61, 911–929.
- Liegl, R.; Wertheimer, C.; Kernt, M.; Docheva, D.; Kampik, A.; Eibl-Lindner, K.H. Attenuation of human lens epithelial cell spreading, migration and contraction via downregulation of the PI3K/Akt pathway. Graefes. Arch. Clin. Exp. Ophthalmol. 2014, 252, 285–292.
- Garcia, B.G.; Wei, Y.; Moron, J.A.; Lin, R.Z.; Javitch, J.A.; Galli, A. Akt is essential for insulin modulation of amphetamine-induced human dopamine transporter cell-surface redistribution. Mol. Pharmacol. 2005, 68, 102–109.
- Wei, Y.; Williams, J.M.; Dipace, C.; Sung, U.; Javitch, J.A.; Galli, A.; Saunders, C. Dopamine transporter activity mediates amphetamine-induced inhibition of Akt through a Ca2+/calmodulin-dependent kinase II-dependent mechanism. Mol. Pharmacol. 2007, 71, 835–842.
- Bourque, M.; Liu, B.; Dluzen, D.E.; di Paolo, T. Sex differences in methamphetamine toxicity in mice: Effect on brain dopamine signaling pathways. Psychoneuroendocrinology 2011, 36, 955–969.
- Ugrumov, M.V. Non-dopaminergic neurons partly expressing dopaminergic phenotype: Distribution in the brain, development and functional significance. J. Chem. Neuroanat. 2009, 38, 241–256.
- Lute, B.J.; Khoshbouei, H.; Saunders, C.; Sen, N.; Lin, R.Z.; Javitch, J.A.; Galli, A. PI3K signaling supports amphetamine-induced dopamine efflux. Biochem. Biophys. Res. Commun. 2008, 372, 656–661.
- Carvelli, L.; Morón, J.A.; Kahlig, K.M.; Ferrer, J.V.; Sen, N.; Lechleiter, J.D.; Leeb-Lundberg, L.M.; Merrill, G.; Lafer, E.M.; Ballou, L.M.; et al. PI 3-kinase regulation of dopamine uptake. J. Neurochem. 2002, 81, 859–869.
- Woodgett, J.R. Recent advances in the protein kinase B signaling pathway. Curr. Opin. Cell Biol. 2005, 17, 150–157.
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603.
- Khattak, M.N.; Buchfelder, M.; Kleindienst, A.; Schöfl, C.; Kremenevskaja, N. CRH and SRIF have opposite effects on the Wnt/β-catenin signalling pathway through PKA/GSK-3β in corticotroph pituitary cells. Cancer Investig. 2010, 28, 797–805.
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426.
- Liang, M.H.; Chuang, D.M. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J. Biol. Chem. 2007, 282, 3904–3917.
- Tian, N.; Kanno, T.; Jin, Y.; Nishizaki, T. Lithium potentiates GSK-3β activity by inhibiting phosphoinositide 3-kinase-mediated Akt phosphorylation. Biochem. Biophys. Res. Commun. 2014, 450, 746–749.
- Liang, M.H.; Wendland, J.R.; Chuang, D.M. Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol. Cell Neurosci. 2008, 37, 440–453.
- Kirshenboim, N.; Plotkin, B.; Shlomo, S.B.; Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Lithium-mediated phosphorylation of glycogen synthase kinase-3β involves PI3 kinase-dependent activation of protein kinase C-α. J. Mol. Neurosci. 2004, 24, 237–245.
- O’Driscoll, C.; Wallace, D.; Cotter, T.G. bFGF promotes photoreceptor cell survival in vitro by PKA-mediated inactivation of glycogen synthase kinase 3β and CREB-dependent Bcl-2 up-regulation. J. Neurochem. 2007, 103, 860–870.
- Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Ryu, J.; Choi, J.; Kim, J.S.; Cho, Y.W.; Roh, G.S.; Kim, H.J.; Cho, G.J.; et al. Caffeine inhibits cell proliferation and regulates PKA/GSK3β pathways in U87MG human glioma cells. Mol. Cells 2011, 31, 275–279.
- Nijholt, I.M.; Dolga, A.M.; Ostroveanu, A.; Luiten, P.G.; Schmidt, M.; Eisel, U.L. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal. 2008, 20, 1715–1724.
- De Joussineau, C.; Sahut-Barnola, I.; Tissier, F.; Dumontet, T.; Drelon, C.; Batisse-Lignier, M.; Tauveron, I.; Pointud, J.C.; Lefrançois-Martinez, A.M.; Stratakis, C.A.; et al. mTOR pathway is activated by PKA in adrenocortical cells and participates in vivo to apoptosis resistance in primary pigmented nodular adrenocortical disease (PPNAD). Hum. Mol. Genet. 2014, 23, 5418–5428.
- Jülich, K.; Sahin, M. Mechanism-based treatment in tuberous sclerosis complex. Pediatr. Neurol. 2014, 50, 290–296.
- Carson, R.P.; Fu, C.; Winzenburger, P.; Ess, K.C. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet. 2013, 22, 140–152.
- Pringle, D.R.; Vasko, V.V.; Yu, L.; Manchanda, P.K.; Lee, A.A.; Zhang, X.; Kirschner, J.M.; Parlow, A.F.; Saji, M.; Jarjoura, D.; et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014, 99, E804–E812.
- McDougle, C.J.; Naylor, S.T.; Cohen, D.J.; Aghajanian, G.K.; Heninger, G.R.; Price, L.H. Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 1996, 53, 993–1000.
- Penedo, L.A.; Oliveira-Silva, P.; Gonzalez, E.M.; Maciel, R.; Jurgilas, P.B.; Melibeu Ada, C.; Campello-Costa, P.; Serfaty, C.A. Nutritional tryptophan restriction impairs plasticity of retinotectal axons during the critical period. Exp. Neurol. 2009, 217, 108–115.
- Schmid, C.L.; Streicher, J.M.; Meltzer, H.Y.; Bohn, L.M. Clozapine acts as an agonist at serotonin 2A receptors to counter MK-801-induced behaviors through a β-arrestin2-independent activation of Akt. Neuropsychopharmacology 2014, 39, 1902–1913.
- Spivak, B.; Golubchik, P.; Mozes, T.; Vered, Y.; Nechmad, A.; Weizman, A.; Strous, R.D. Low platelet-poor plasma levels of serotonin in adult autistic patients. Neuropsychobiology 2004, 50, 157–160.
- Cross, S.; Kim, S.J.; Weiss, L.A.; Delahanty, R.J.; Sutcliffe, J.S.; Leventhal, B.L.; Cook, E.H., Jr.; Veenstra-Vanderweele, J. Molecular genetics of the platelet serotonin system in first-degree relatives of patients with autism. Neuropsychopharmacology 2008, 33, 353–360.
- Iwata, K.; Matsuzaki, H.; Tachibana, T.; Ohno, K.; Yoshimura, S.; Takamura, H.; Yamada, K.; Matsuzaki, S.; Nakamura, K.; Tsuchiya, K.J.; et al. N-ethylmaleimide-sensitive factor interacts with the serotonin transporter and modulates its trafficking: Implications for pathophysiology in autism. Mol. Autism 2014, 5, 33.
- Prasad, H.C.; Steiner, J.A.; Sutcliffe, J.S.; Blakely, R.D. Enhanced activity of human serotonin transporter variants associated with autism. Philos. Trans. R. Soc. Lond. B 2009, 364, 163–173.
- Aggarwal, B.B.; Yuan, W.; Li, S.; Gupta, S.C. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: Identification of novel components of turmeric. Mol. Nutr. Food Res. 2013, 57, 1529–1542.
- Wang, R.; Li, Y.H.; Xu, Y.; Li, Y.B.; Wu, H.L.; Guo, H.; Zhang, J.Z.; Zhang, J.J.; Pan, X.Y.; Li, X.J. Curcumin produces neuroprotective effects via activating brain-derived neurotrophic factor/TrkB-dependent MAPK and PI-3K cascades in rodent cortical neurons. Prog Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 147–153.
- Wu, J.; Li, Q.; Wang, X.; Yu, S.; Li, L.; Wu, X.; Chen, Y.; Zhao, J.; Zhao, Y. Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS One 2013, 8, e59843.
- Lipecka, J.; Norez, C.; Bensalem, N.; Baudouin-Legros, M.; Planelles, G.; Becq, F.; Edelman, A.; Davezac, N. Rescue of ΔF508-CFTR (cystic fibrosis transmembrane conductance regulator) by curcumin: Involvement of the keratin 18 network. J. Pharmacol. Exp. Ther. 2006, 317, 500–505.
- Yu, Y.C.; Miki, H.; Nakamura, Y.; Hanyuda, A.; Matsuzaki, Y.; Abe, Y.; Yasui, M.; Tanaka, K.; Hwang, T.C.; Bompadre, S.G.; et al. Curcumin and genistein additively potentiate G551D-CFTR. J. Cyst. Fibros. 2011, 10, 243–252.
- Zhang, C.; Browne, A.; Child, D.; Divito, J.R.; Stevenson, J.A.; Tanzi, R.E. Loss of function of ATXN1 increases amyloid β-protein levels by potentiating β-secretase processing of β-amyloid precursor protein. J. Biol. Chem. 2010, 285, 8515–8526.
- Benammi, H.; el Hiba, O.; Romane, A.; Gamrani, H. A blunted anxiolytic like effect of curcumin against acute lead induced anxiety in rat: Involvement of serotonin. Acta Histochem. 2014, 116, 920–925.
- Burgess, J.R.; Stevens, L.; Zhang, W.; Peck, L. Long-chain polyunsaturated fatty acids in children with attention-deficit hyperactivity disorder. Am. J. Clin. Nutr. 2000, 71, 327S–330S.
- Montgomery, P.; Burton, J.R.; Sewell, R.P.; Spreckelsen, T.F.; Richardson, A.J. Low blood long chain ω-3 fatty acids in UK children are associated with poor cognitive performance and behavior: A cross-sectional analysis from the DOLAB study. PLoS One 2013, 8, e66697.
- Jia, D.; Heng, L.J.; Yang, R.H.; Gao, G.D. Fish oil improves learning impairments of diabetic rats by blocking PI3K/AKT/nuclear factor-κB-mediated inflammatory pathways. Neuroscience 2014, 258, 228–237.
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 35.
- Kitagishi, Y.; Matsuda, S. Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers Res. Ther. 2013, 5, 42.
- Xie, F.; Su, M.; Qiu, W.; Zhang, M.; Guo, Z.; Su, B.; Liu, J.; Li, X.; Zhou, L. Kaempferol promotes apoptosis in human bladder cancer cells by inducing the tumor suppressor, PTEN. Int. J. Mol. Sci. 2013, 14, 21215–21226.
- Gao, N.; Budhraja, A.; Cheng, S.; Liu, E.H.; Chen, J.; Yang, Z.; Chen, D.; Zhang, Z.; Shi, X. Phenethyl isothiocyanate exhibits antileukemic activity in vitro and in vivo by inactivation of Akt and activation of JNK pathways. Cell Death Dis. 2011, 2, e140.


