Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yongjun Chen | -- | 3236 | 2023-09-05 19:05:15 | | | |
| 2 | Rita Xu | -29 word(s) | 3207 | 2023-09-06 03:20:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, Q.; Xiong, F.; Wu, G.; Wang, D.; Liu, W.; Chen, J.; Qi, Y.; Wang, B.; Chen, Y. SMAD Proteins in TGF-β Signalling Pathway in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/48848 (accessed on 27 July 2026).
Wang Q, Xiong F, Wu G, Wang D, Liu W, Chen J, et al. SMAD Proteins in TGF-β Signalling Pathway in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/48848. Accessed July 27, 2026.
Wang, Qi, Fei Xiong, Guanhua Wu, Da Wang, Wenzheng Liu, Junsheng Chen, Yongqiang Qi, Bing Wang, Yongjun Chen. "SMAD Proteins in TGF-β Signalling Pathway in Cancer" Encyclopedia, https://encyclopedia.pub/entry/48848 (accessed July 27, 2026).
Wang, Q., Xiong, F., Wu, G., Wang, D., Liu, W., Chen, J., Qi, Y., Wang, B., & Chen, Y. (2023, September 05). SMAD Proteins in TGF-β Signalling Pathway in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/48848
Wang, Qi, et al. "SMAD Proteins in TGF-β Signalling Pathway in Cancer." Encyclopedia. Web. 05 September, 2023.
Copy Citation
Suppressor of mother against decapentaplegic (SMAD) family proteins are central to one of the most versatile cytokine signalling pathways in metazoan biology, the transforming growth factor-β (TGF-β) pathway. The TGF-β pathway is widely known for its dual role in cancer progression as both an inhibitor of tumour cell growth and an inducer of tumour metastasis.
SMAD
transforming growth factor-β
cancer
1. Introduction
Modern medicine shows that the development of tumours is a very complex process. It is the combined result of tumour cell proliferation, metastasis, and apoptosis. A variety of growth factors and signalling proteins are involved in this process. Members of the TGF-β family control cell growth, differentiation, and apoptosis, and have important functions during embryonic development [1]. In tumours, multiple cell types produce TGF-β and respond to it, resulting in a complex network involving epithelial cells, tumour cells, immune cells, and stromal fibroblasts [2]. This complex network can cause disease and change over time, giving TGF-β both tumour suppressive and tumour promoting or enabling effects [3]. In normal non-cancerous and early cancer cells, activation of the TGF-β signalling pathway induces apoptosis and strongly inhibits the cell cycle [4][5], suggesting a major role for this signalling pathway in cancer suppression. On the other hand, the TGF-β pathway exerts tumour-promoting effects by activating multiple signalling pathways and by affecting EMT, angiogenesis and tumour invasion, and metastasis through immune evasion [6]. The key molecules that enable the TGF-β pathway to play different roles are the SMAD proteins.
The first protein of the SMAD family, an intracellular protein named Mad, was identified in the 1990s during a mutation screen for the decapentaplegic (dpp) gene responsible for the formation of Drosophila wings [7]. Subsequently, homologues of Mad proteins were identified in Caenorhabditis elegans and vertebrates. The term “SMAD” is derived from the combination of the gene names of two homologous proteins, Sma of Caenorhabditis elegans and Mad of Drosophila melanogaster [8]. The first SMAD to be discovered was SMAD4 or DPC4 (deleted in pancreatic cancer 4) [9].
Eight SMAD proteins are encoded by human and mouse genomes. Of these, SMAD1, SMAD2, SMAD3, SMAD5, and SMAD8 act as substrates for the TGF-β family receptor, and these proteins are commonly referred to as receptor-regulated SMADs (R-SMADs). SMAD1, 5, and 8 are mainly substrates for bone morphogenetic proteins (BMPs) and anti-Mullerian receptors; SMAD2 and 3 are principally substrates for the TGFβ, activin, and Nodal receptors. SMAD4, also known as Co-SMAD, is a cofactor for all R-SMADs. SMAD6 and 7 are inhibitory SMADs (I-SMADs) that have antagonistic effects and eliminate TGF-β signalling [10][11]. SMAD6 preferentially inhibits BMP signalling, whereas SMAD7 inhibits both the TGF-β/activin and BMP signalling pathways [12].
Structurally, the SMAD proteins consist of two spherical structural domains: the N-terminal MAD Homology domain1 (MH1), which contains a hairpin 4 structure with DNA binding capacity, and the C-terminal MAD Homology domain2 (MH2), which contains a hydrophobic element that binds to the transmembrane receptors TGF-βR and BMPR. The intermediate linker region is a flexible fragment with post-translational modification sites such as the binding site for SMURF (SMAD ubiquitination-related factor) ubiquitin ligase and the phosphorylation site for protein kinase such as TGF-β type I receptor [10].
SMAD family proteins function primarily in the TGF-β pathway. TGF-β family ligands initiate downstream signalling events by activating transmembrane serine-threonine kinase receptors, namely, the TGF-β type I receptor (TbRI), which is also named as activin receptor-like kinase 5 (ALK5), and the TGF-β type II receptor (TbRII). On ligand binding, TbRII forms a heterotetramer with TbRI/ALK5, and the glycine/serine-rich (GS) structural domain of TbRI/ALK5 is phosphorylated by TbRII, which is a 30 amino acid region rich in glycine and serine that precedes the kinase structural domain. Anchoring proteins capture R-SMADs for presentation to activated type I receptors [13][14]. Inactive SMADs are in an inhibitory state through intermolecular interactions between the MH1 and MH2 structural domains [15]. The MH2 structural domain of R-SMADs is phosphorylated in the presence of activated type I receptors, thereby relieving their inhibitory state. Activated R-SMADs bind to Co-SMAD and SMAD4 to form a polymer that completes the SMAD4-mediated plasmid-nucleus shuttle. Because of the low binding affinity of the SMAD complex for DNA, other transcription factors are required for high-affinity interactions and chromatin specificity. After entering the nucleus, the SMAD complex recruits co-activators such as P300/CBP [16][17] and GCN5 [18] to exert transcriptional activation. At the same time, it can also recruit co-repressors such as TGIF [19], Ski [20][21], and SnoN [22] to exert transcriptional repression. Meanwhile, the I-SMADs, especially SMAD7, inhibit SMAD-mediated signalling by competing with activated R-SMADs to bind SMAD4 or interacting with upstream receptors (Figure 1) [23].
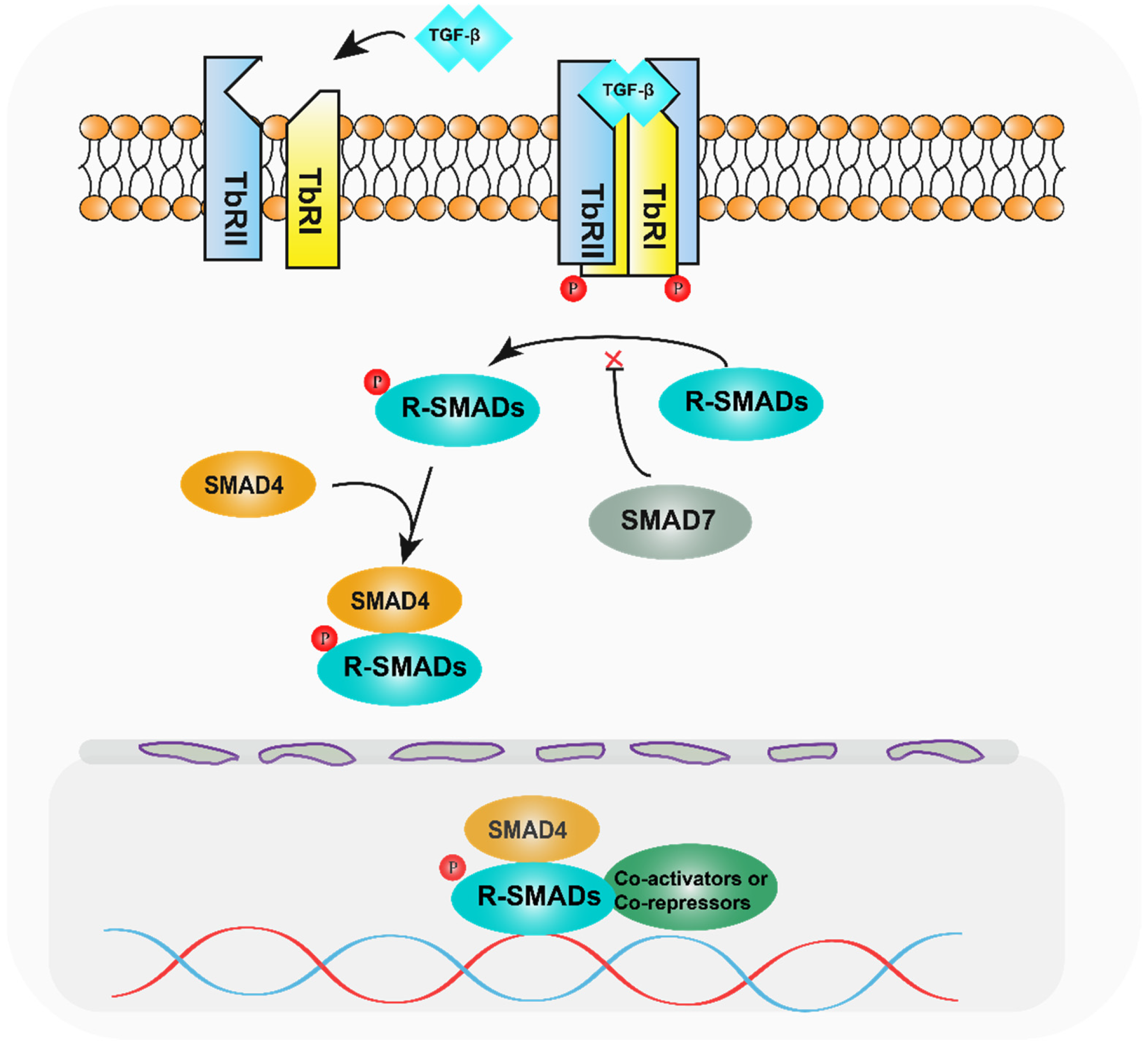
Figure 1. Diagram of the TGF-β pathway mechanism. After TGF-β activates the relevant receptors, the receptors phosphorylate R-SMADs, which bind to SMAD4 and enter the nucleus, recruiting relevant transcription factors and regulating target genes.
The dual role of TGF-β in cancer has long been identified, but its mechanistic basis, operational logic, and clinical relevance are still unclear. Researchers summarize the recent progress in the research and application of SMAD proteins in tumours, in order to find the contribution of SMAD proteins to the dual role of TGF-β and provide clues for the subsequent research on SMAD proteins in tumours.
2. Oesophageal Cancer
Barrett’s oesophagus (BE) is formed when squamous epithelium is replaced by precancerous tissue (dysplasia) and is associated with an increased risk of developing oesophageal adenocarcinoma (EAC) [24]. Studies have shown that SMAD family proteins and the TGF-β signalling pathway play an important role in the progression of Barrett’s oesophagus to oesophageal cancer.
The percentage of linker threonine-phosphorylated SMAD2/3 (pSMAD2/3L-Thr) positive cells increases significantly with the progression of oesophageal tumours. In low-grade intraepithelial tumours, pSMAD2/3L-Thr-positive cells were distributed in the lower segment of low-grade intraepithelial neoplasia, and were observed up to the upper section in carcinoma in situ. In invasive squamous cell carcinoma, they were scattered throughout the tumour with a loss of polarity and were found in primary intraepithelial lesions and at sites of submucosal and vascular invasion. pSMAD2/3L-Thr was significantly expressed in human oesophageal non-tumour and tumour epithelium, suggesting that they are epithelial and cancer stem cells associated with oesophageal tumourigenesis, respectively [25]. Furthermore, SMAD4 expression was significantly reduced in all stages of Barrett’s oesophagus from chemotaxis to hypo- and highly heterogeneous hyperplasia [26], and knockdown of SMAD4 was sufficient to promote tumourigenesis in dysplastic Barrett’s oesophagus cells in vivo [27]. Heterozygous deletions of the region on chromosome 18q containing SMAD2 and SMAD4 were present in approximately 30–70% of patients with precancerous BE [28] and in approximately 70% of oesophageal adenocarcinomas associated with BE [28][29], suggesting that such heterozygous deletions are early events in tumour transformation.
In oesophageal cancer, ATP-binding cassette, sub-family B (MDR/TAP), member 7 (ABCB7) knockdown inhibits TGF-β signalling pathway transduction by promoting SMAD7 expression and repressing SMAD3 expression, inducing apoptosis and suppressing EMT in oesophageal cancer cells [30]. The transcription factor forkhead box D3 (FOXD3) can bind directly to the promoter region of the SMAD7 gene, leading to the transcriptional promotion of SMAD7 in human oesophageal cancer cells, inhibition of the TGF-β pathway, and suppressive effects in oesophageal squamous cell carcinoma [31]. It was shown that SMAD7, as a repressor molecule of the TGF-β pathway, is underexpressed in oesophageal carcinoma through a variety of genes, allowing for abnormal activation of the TGF-β pathway and promoting the progression of oesophageal carcinoma.
3. Gastric Cancer
Gastric cancer is one of the most common malignant tumours in the world and has a high morbidity and mortality rate. Although significant progress has been made in the treatment of gastric cancer, the 5-year survival rate is only 20–30% due to factors such as the lack of specific clinical manifestations and targeted drugs in the early stages [32].
In gastric cancer, ubiquitin-related enzymes such as TRIM22 [33] and USP32 [34] promote the proliferation and invasion of gastric cancer cells by affecting the stability of the SMAD2 protein, which is highly expressed in gastric cancer. SMAD2 expression levels are low in EBV-associated gastric cancer, particularly in the presence of EBV-encoded latent membrane protein 2A (LMP2A). It has been shown that LMP2A promotes miR-155-5p expression through the activation of nuclear factor-κB (NFκB) signalling, and the overexpression of miR-155-5p inhibits SMAD2. Immunofluorescence analysis further showed that LMP2A blocks p-SMAD2 translocation to the nucleus. Thus, the role of LMP2A in EBV-positive GC may lead to a favourable prognosis by promoting apoptosis and cell cycle arrest as well as inhibiting tumour proliferation [35]. The above studies suggest that SMAD2, an important mediator of the TGF-β pathway, plays an important role as an oncogene in the development of gastric cancer.
In terms of tumour treatment, different treatment strategies are associated with SMAD expression. Surgery alone increased the expression levels of SMAD1, SMAD2, and SMAD4. Conversely, treatment with 5-FU-based adjuvants decreased the expression levels of SMAD3 and SMAD6, but increased the expression of SMAD5. In addition, high levels of SMAD9 expression were associated with adverse effects in patients treated with other adjuvants [36]. The expression levels of SMAD proteins have an important impact on the tumour microenvironment and therapeutic efficacy. Studies have shown that SMAD1 is highly expressed in cisplatin-resistant gastric cancer cells and that SMAD1 interacts with YAP1, leading to the increased resistance of gastric cancer cells to cisplatin [37].
The introduction of immunotherapy, represented by immune checkpoint blockade, has brought about a turnaround in the treatment of gastric cancer. However, clinical studies have found that the efficacy of this treatment varies greatly between individual patients. Recently, the impact of SMAD family protein expression levels on immunotherapy has been increasingly recognised. It was shown that SMAD4 deletion allowed gastric cancer cells to evade tumour immunity. SMAD4-deficient GC cells exhibited the expansion of CD133+ cancer stem cells, along with the inhibition of dendritic cell (DC) differentiation and the aggregation of cytotoxic T cells with granulocyte myeloid-derived suppressor cells (G-MDSC) via CXCL1-containing secretomes. In addition, SMAD4 deletion increased programmed cell death ligand-1 (PD-L1) and decreased 4-1BBL expression, indicating altered immunogenicity. Combined immune checkpoint blockade (ICB) with anti-PD-L1 and anti-CTLA-4 or agonistic anti-4-1BB antibodies effectively treats ICB monotherapy-resistant SMAD4-deficient allografts, providing a rational basis for an ICB strategy to treat advanced SMAD4-deficient GC [38].
Due to the important role of SMAD family proteins in the development of gastric cancer and response to treatment, SAMD protein levels are closely related to the prognosis of gastric cancer patients. Studies have shown that higher levels of SMAD1, SMAD2, and SMAD4 expression are associated with good overall survival (OS) in stage I and II cancer. On the other hand, increased expression of SMAD3, SMAD5, SMAD6, and SMAD7 was associated with low OS in stage I and II of the cancer. In all gastric cancers, increased SMAD9 expression was associated with poor OS [36]. In a subgroup analysis based on tumour node metastasis (TNM) stage, SMAD4 and SMAD7 showed the most significant prognostic differences in patients with stage I gastric cancer [39]. Based on previous studies, it is known that the TGF-β pathway promotes the EMT process in tumours and is often associated with a poorer prognosis. However, the above data suggest that activation of the TGF-β pathway improves the prognosis of some patients. Therefore, the complexity of whether TGF-β signalling is tumour-suppressive or tumour-promoting remains to be further investigated.
4. Colorectal Cancer
The role of SMAD proteins in colorectal cancer is more complex and well-studied. In CRC development, as early as 1998, Zhu et al. found that mice with a full knockout of SMAD3 could spontaneously develop CRC [40]. More recently, Gu et al. showed that mice heterozygous for SMAD4 and the gene encoding the SMAD3 adaptor protein SPTBN1 also developed CRC spontaneously and that these mice exhibited altered gut microbiota that resembled that associated with human CRC [41]. Furthermore, BRAF-driven colorectal cancer is one of the most poorly prognosed subtypes of colorectal cancer. In the oncogenic BRAF-V600E mouse model, deletion of the tumour suppressor SMAD4 promoted rapid development and the progression of serrated tumours, and SMAD4 mutations co-occurred with BRAF-V600E mutations in human patient tumours [42]. These findings identify SMAD4 as a key factor in early-stage serrated cancers and help to expand the understanding of this rare but aggressive subgroup of colorectal cancers.
For the progression of CRC, SMAD4 plays an important role. A growing body of evidence confirms that SMAD4 is lost in colorectal cancer at a frequency of approximately 30% [43]. Inactivation of SMAD4 leads to increased secretion of a variety of proteins known to be involved in pro-metastatic processes. For example, it has been shown that DKK3, one of the factors secreted organ-specific by SMAD4 mutants, reduces the antitumour effects of natural killer cells (NK cells) [44]. TRIM47 promotes the expression of CCL15 by promoting SMAD4 ubiquitination and degradation, and promotes the growth and invasion of human CRC cells through CCL15-CCR1 signalling [45]. Furthermore, in colorectal cancer, the ribosomal biogenesis factor NLE1 plays a key role in tumour growth and progression. In the absence of SMAD4, the TGF-β signalling pathway-mediated downregulation of NLE1 is prevented by the ectopic expression of c-MYC, which occupies the E-box-containing region within the NLE1 promoter and upregulates NLE1, thereby promoting tumour progression [46]. The above studies suggest that decreased expression of SMAD4 has a facilitative effect on CRC progression.
However, SMAD4 does not completely inhibit CRC progression. Li et al. found that SMAD4 in combination with SMAD3 could positively regulate the vascular endothelial growth factor C (VEGF-C) during colon cancer metastasis by binding to the promoter of the VEGF-C gene, which is essential for invasive metastasis in CRC. However, at the same time, SMAD4 increased the transcription of miR-128-3p, a microRNA targeting VEGF-C mRNA, leading to the downregulation of VEGF-C expression. Ultimately, Li et al. found that the long non-coding RNA (lncRNA) ASLNC07322, which is specifically increased in metastatic colon cancer, acts as a sponge for miR-128-3p to reduce it, leading to a subsequent increase in VEGF-C. ASLNC07322 critically controls this negative and positive regulatory transition between them, which in turn balances the SMAD4 effects on CRC invasive metastasis [47].
The role of the SMAD family of proteins on the immune microenvironment of tumours and the tumour immune response has also received considerable attention in recent years. Hanna et al. found that the regulation of mucosal inflammation in ulcerative colitis-associated tumours was central to the tumour suppressive function of SMAD4 in the colon. SMAD4 deficiency in the mouse colonic epithelium leads to an expansion of gut-associated lymphoid tissue and the recruitment of immune cells to the mouse colonic epithelium and stroma, particularly T regulatory cells, Th17, and dendritic cells. A key downstream node of this regulation is the inhibition of epithelial chemokine c-c motif chemokine ligand 20 (CCL20) signalling to CCL20/c-c motif chemokine receptor 6 (CCR6) in immune cells. Deletion of SMAD4 in colonic epithelial cells increases CCL20 expression and chemotaxis of CCR6+ immune cells, contributing to increased susceptibility to colon cancer [48]. In addition, interferon-γ cell expression was significantly increased in T cells and colonic mucosal epithelium of mice deficient in SMAD4, and increased IFN-γ expression promoted colorectal carcinogenesis through immunomodulatory mechanisms and directly on endothelial and epithelial homeostasis. SMAD4 knockdown also upregulated CXCL1 and CXCL8 expression, recruiting neutrophils into colorectal tumours. Both CXCL1 and CXCL8 were abundantly expressed in tumour-infiltrating neutrophils. Statistical analysis showed that CRC patients with high levels of CXCL8 exhibited shorter OS and recurrence-free survival (RFS) [49]. The above studies suggest that SMAD4 deficiency contributes to colorectal cancer development by affecting the response of immune cells.
Given the important role of SMAD4 in the development of CRC, SMAD4 has a significant indicative role in the prognosis of CRC patients. Analysis of a cohort of 364 patients with stage I–IV CRC showed that SMAD4 deficiency was associated with higher tumour and nodal staging, the use of adjuvant therapy, fewer tumour infiltrating lymphocytes (TIL), lower peritumour lymphocyte aggregation (PLA) scores, and poorer RFS. Among patients receiving 5-fluorouracil (5-FU)-based systemic chemotherapy, the median RFS was 3.8 years in SMAD4-deficient patients compared to 13 years in SMAD4-preserved patients [50]. In conclusion, SMAD4 deficiency was associated with poorer clinical prognosis, chemoresistance, and reduced immune infiltration, supporting its use as a prognostic marker in cancer patients. An analysis of the prognostic and predictive value of actionable mutated genes in metastatic colorectal cancer (mCRC) revealed that concurrent mutations in TP53 and SMAD4 were associated with an increased risk of death (p = 0.03; HR:2.91). In mCRC patients treated with first-line regimens, SMAD4 mutations in TP53-altered tumours predicted negative prognostic outcomes [51]. In addition, mutational status of SMAD4 had a role in predicting prognosis after resection of liver metastases from colorectal cancer [52].
For the diagnosis of CRC, SMAD3 has a more prominent role. In a Taiwanese cohort study, investigators found hypomethylated SMAD3 in 91.4% (501/548) of Taiwanese colorectal cancer tissues and 66.6% of benign tubular adenoma-polyp tissues. In addition, SMAD3 hypomethylation was observed in 94.7% of CRC patients in the Cancer Genome Atlas dataset. A reduction in the methylation of SMAD3 in circulating cell-free was detected in 70% of CRC patients, but in only 20% of healthy individuals. SMAD3 mRNA expression was low in 42.9% of Taiwanese CRC tumour tissues but high in 29.4% of tumours compared with paired adjacent normal tissues. These results suggest that SMAD3 hypermethylation is a potential diagnostic marker for CRC in Western and Asian populations [53]. Another study found that the microRNA-375 and rs4939827 SNPs in SMAD7 could be considered as potential markers for the detection and early diagnosis of CRC patients [54].
SMAD3 is also a key molecule in the chemoresistant phenotype of CRC. In a group of 76 patients with locally advanced rectal cancer (LARC), the expression of SMAD3 and phosphorylated SMAD3 in preoperative tumour tissues was assessed by immunohistochemistry, and SMAD3 polymorphisms (rs35874463, rs1065080, rs1061427, rs17228212, rs744910, and rs745103) in relation to tumour regression grade and patient prognosis. The results showed that patients with high tumour expression of SMAD3 were at significantly increased risk of poor response to neoadjuvant chemotherapy. Patients carrying the variant SMAD3 rs745103-G allele had a slightly poorer response (OR:0.48, p = 0.0093), longer OS (HR:0.65, p = 0.0307), and a trend towards prolonged progression-free survival (HR:0.75, p = 0.0944). Patients carrying both high SMAD3 tumour expression and the wild-type rs745103-A allele had an extremely high risk of failing to achieve a complete response (OR:13.45, p = 0.0005). Host and tumour SMAD3 status may be considered to improve the risk stratification of LARC patients to facilitate the selection of other personalised neoadjuvant treatment strategies including intensive treatment regimens [55]. In addition, Mattia et al. found that patients with the SMAD3rs7179840-C allele present in CRC tumours had a higher OS rate after treatment with FOLFIRI (irinotecan, 5-FU, folinic acid). This finding may provide a new decision-making tool for improving the clinical management of CRC patients receiving FOLFIRI [56].
For SMAD7, which has a dual role in CRC progression, it promotes tumourigenicity in non-tumourigenic CRC cell lines by inhibiting the TGF-β signalling pathway [57]. In addition, CRC cells contain high levels of active signal transducers and transcriptional activators (STAT)-3 that exert proliferative and anti-apoptotic effects. There is a positive correlation between SMAD7 and STAT3, promoting STAT3 expression and acting as a promoter of CPC progression [58]. In contrast, the m1 A demethylase alkB homolog 1 (ALKBH1) promoted CRC metastasis by promoting methyltransferase 3, N6-adenosine-methyltransferase (METTL3), which destabilised SMAD7 [59]. This suggests that SMAD7 plays an inhibitory role in CRC progression. In conclusion, the SMAD protein family, which provides important indications for the early diagnosis of CRC and the choice of treatment options, offers great clinical value.
References
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873.
- Bierie, B.; Moses, H.L. TGF-β and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40.
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71.
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369.
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230.
- Ali, S.; Rehman, M.U.; Yatoo, A.M.; Arafah, A.; Khan, A.; Rashid, S.; Majid, S.; Ali, A.; Ali, M.N. TGF-β signaling pathway: Therapeutic targeting and potential for anti-cancer immunity. Eur. J. Pharmacol. 2023, 947, 175678.
- Sekelsky, J.J.; Newfeld, S.J.; Raftery, L.A.; Chartoff, E.H.; Gelbart, W.M. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 1995, 139, 1347–1358.
- Derynck, R.; Gelbart, W.M.; Harland, R.M.; Heldin, C.H.; Kern, S.E.; Massagué, J.; Melton, D.A.; Mlodzik, M.; Padgett, R.W.; Roberts, A.B.; et al. Nomenclature: Vertebrate mediators of TGFβ family signals. Cell 1996, 87, 173.
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353.
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700.
- Cook, T.; Urrutia, R. TIEG proteins join the Smads as TGF-β-regulated transcription factors that control pancreatic cell growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G513–G521.
- Itoh, S.; Landstrom, M.; Hermansson, A.; Itoh, F.; Heldin, C.H.; Heldin, N.E.; Dijke, P.T. Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 1998, 273, 29195–29201.
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347.
- Massagué, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791.
- Hata, A.; Lo, R.S.; Wotton, D.; Lagna, G.; Massagué, J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature 1997, 388, 82–87.
- Feng, X.H.; Zhang, Y.; Wu, R.Y.; Derynck, R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-β-induced transcriptional activation. Genes Dev. 1998, 12, 2153–2163.
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-β-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119.
- Kahata, K.; Hayashi, M.; Asaka, M.; Hellman, U.; Kitagawa, H.; Yanagisawa, J.; Kato, S.; Imamura, T.; Miyazono, K. Regulation of transforming growth factor-β and bone morphogenetic protein signalling by transcriptional coactivator GCN5. Genes Cells 2004, 9, 143–151.
- Wotton, D.; Lo, R.S.; Lee, S.; Massague, J. A Smad transcriptional corepressor. Cell 1999, 97, 29–39.
- Akiyoshi, S.; Inoue, H.; Hanai, J.; Kusanagi, K.; Nemoto, N.; Miyazono, K.; Kawabata, M. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with smads. J. Biol. Chem. 1999, 274, 35269–35277.
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev. 1999, 13, 2196–2206.
- Sun, Y.; Liu, X.; Eaton, E.N.; Lane, W.S.; Lodish, H.F.; Weinberg, R.A. Interaction of the Ski oncoprotein with Smad3 regulates TGF-β signaling. Mol. Cell 1999, 4, 499–509.
- Itoh, F.; Asao, H.; Sugamura, K.; Heldin, C.H.; Dijke, P.T.; Itoh, S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. 2001, 20, 4132–4142.
- Rastogi, A.; Puli, S.; El-Serag, H.B.; Bansal, A.; Wani, S.; Sharma, P. Incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus and high-grade dysplasia: A meta-analysis. Gastrointest. Endosc. 2008, 67, 394–398.
- Horitani, S.; Fukui, T.; Tanimura, Y.; Matsumoto, Y.; Miyamoto, S.; Tanaka, T.; Tomiyama, T.; Ikeura, T.; Ando, Y.; Nishio, A.; et al. Specific Smad2/3 Linker Phosphorylation Indicates Esophageal Non-neoplastic and Neoplastic Stem-Like Cells and Neoplastic Development. Dig. Dis. Sci. 2021, 66, 1862–1874.
- Onwuegbusi, B.A.; Aitchison, A.; Chin, S.F.; Kranjac, T.; Mills, I.; Huang, Y.; Lao-Sirieix, P.; Caldas, C.; Fitzgerald, R.C. Impaired transforming growth factor β signalling in Barrett’s carcinogenesis due to frequent SMAD4 inactivation. Gut 2006, 55, 764–774.
- Gotovac, J.R.; Kader, T.; Milne, J.V.; Fujihara, K.M.; Lara-Gonzalez, L.E.; Gorringe, K.L.; Kalimuthu, S.N.; Jayawardana, M.W.; Duong, C.P.; Phillips, W.A.; et al. Loss of SMAD4 Is Sufficient to Promote Tumorigenesis in a Model of Dysplastic Barrett’s Esophagus. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 689–713.
- Wu, T.T.; Watanabe, T.; Heitmiller, R.; Zahurak, M.; Forastiere, A.A.; Hamilton, S.R. Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am. J. Pathol. 1998, 153, 287–294.
- Barrett, M.T.; Schutte, M.; Kern, S.E.; Reid, B.J. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res. 1996, 56, 4351–4353.
- Feng, S.; Jia, J.; Lv, G.; Wang, Y. Knockdown of ABCB7 inhibits esophageal cancer progression by inhibiting the TGF-β/Smad signaling. Arch. Biochem. Biophys. 2023, 742, 109620.
- Wu, Z.; Li, Y.; Niu, Y.; Lu, J.; Yan, Z.; Xu, T.; Guo, Y.; Dong, Z.; Guo, W. FOXD3 suppresses epithelial-mesenchymal transition through direct transcriptional promotion of SMAD7 in esophageal squamous cell carcinoma. Mol. Carcinog. 2021, 60, 859–873.
- Suzuki, H.; Oda, I.; Abe, S.; Sekiguchi, M.; Mori, G.; Nonaka, S.; Yoshinaga, S.; Saito, Y. High rate of 5-year survival among patients with early gastric cancer undergoing curative endoscopic submucosal dissection. Gastric Cancer 2016, 19, 198–205.
- Zhou, Z.; Gao, W.; Yuan, B.; Zhang, S.; Wang, K.; Du, T. TRIM22 inhibits the proliferation of gastric cancer cells through the Smad2 protein. Cell Death Discov. 2021, 7, 234.
- Dou, N.; Hu, Q.; Li, L.; Wu, Q.; Li, Y.; Gao, Y. USP32 promotes tumorigenesis and chemoresistance in gastric carcinoma via upregulation of SMAD2. Int. J. Biol. Sci. 2020, 16, 1648–1657.
- Shi, Q.; Zhang, Y.; Liu, W.; Xiao, H.; Qi, Y.; Li, J.; Luo, B. Latent membrane protein 2A inhibits expression level of Smad2 through regulating miR-155-5p in EBV-associated gastric cancer cell lines. J. Med. Virol. 2020, 92, 96–106.
- Zhang, H.-W.; Guo, Y.; Sun, L.-X.; Ni, F.-B.; Xu, K. Prognostic value of small mother against decapentaplegic expression in human gastric cancer. Bioengineered 2021, 12, 2534–2549.
- Chen, W.; Hu, J.; He, Y.; Yu, L.; Liu, Y.; Cheng, Y.; Jia, B.; Li, X.; Yu, G.; Wang, Y. The Interaction Between SMAD1 and YAP1 Is Correlated with Increased Resistance of Gastric Cancer Cells to Cisplatin. Appl. Biochem. Biotechnol. 2022, 194, 1–18.
- An, H.-W.; Seok, S.H.; Kwon, J.-W.; Choudhury, A.D.; Oh, J.-S.; Voon, D.C.; Kim, D.-Y.; Park, J.W. The loss of epithelial Smad4 drives immune evasion via CXCL1 while displaying vulnerability to combinatorial immunotherapy in gastric cancer. Cell Rep. 2022, 41, 111878.
- Kim, Y.H.; Lee, H.S.; Lee, H.J.; Hur, K.; Kim, W.H.; Bang, Y.J.; Kim, S.J.; Lee, K.U.; Choe, K.J.; Yang, H.K. Prognostic significance of the expression of Smad4 and Smad7 in human gastric carcinomas. Ann. Oncol. 2004, 15, 574–580.
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714.
- Gu, S.; Zaidi, S.; Hassan, M.I.; Mohammad, T.; Malta, T.M.; Noushmehr, H.; Nguyen, B.; Crandall, K.A.; Srivastav, J.; Obias, V.; et al. Mutated CEACAMs Disrupt Transforming Growth Factor Beta Signaling and Alter the Intestinal Microbiome to Promote Colorectal Carcinogenesis. Gastroenterology 2020, 158, 238–252.
- Tong, K.; Kothari, O.A.; Haro, K.S.; Panda, A.; Bandari, M.M.; Carrick, J.N.; Hur, J.J.; Zhang, L.; Chan, C.S.; Xing, J.; et al. SMAD4 is critical in suppression of BRAF-V600E serrated tumorigenesis. Oncogene 2021, 40, 6034–6048.
- Wang, H.; Stephens, B.; Von Hoff, D.D.; Han, H. Identification and characterization of a novel anticancer agent with selectivity against deleted in pancreatic cancer locus 4 (DPC4)-deficient pancreatic and colon cancer cells. Pancreas 2009, 38, 551–557.
- Dijkstra, J.J.; Neikes, H.K.; Rezaeifard, S.; Ma, X.; Voest, E.E.; Tauriello, D.V.F.; Vermeulen, M. Multiomics of Colorectal Cancer Organoids Reveals Putative Mediators of Cancer Progression Resulting from SMAD4 Inactivation. J. Proteome Res. 2023, 22, 138–151.
- Liang, Q.; Tang, C.; Tang, M.; Zhang, Q.; Gao, Y.; Ge, Z. TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J. Exp. Clin. Cancer Res. 2019, 38, 159.
- Loevenich, L.P.; Tschurtschenthaler, M.; Rokavec, M.; Silva, M.G.; Jesinghaus, M.; Kirchner, T.; Klauschen, F.; Saur, D.; Neumann, J.; Hermeking, H.; et al. SMAD4 Loss Induces c-MYC-Mediated NLE1 Upregulation to Support Protein Biosynthesis, Colorectal Cancer Growth, and Metastasis. Cancer Res. 2022, 82, 4604–4623.
- Li, X.; Lv, X.; Li, Z.; Li, C.; Li, X.; Xiao, J.; Liu, B.; Yang, H.; Zhang, Y. Long Noncoding RNA ASLNC07322 Functions in VEGF-C Expression Regulated by Smad4 during Colon Cancer Metastasis. Mol. Ther. Nucleic Acids 2019, 18, 851–862.
- Hanna, D.N.; Smith, P.M.; Novitskiy, S.V.; Washington, M.K.; Zi, J.; Weaver, C.J.; Hamaamen, J.A.; Lewis, K.B.; Zhu, J.; Yang, J.; et al. SMAD4 Suppresses Colitis-associated Carcinoma Through Inhibition of CCL20/CCR6-mediated Inflammation. Gastroenterology 2022, 163, 1334–1350.e14.
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Recruiting Tumor-Associated Neutrophils via the CXCL1/8-CXCR2 Axis. Clin. Cancer Res. 2019, 25, 2887–2899.
- Wasserman, I.; Lee, L.H.; Ogino, S.; Marco, M.R.; Wu, C.; Chen, X.; Datta, J.; Sadot, E.; Szeglin, B.; Guillem, J.G.; et al. SMAD4 Loss in Colorectal Cancer Patients Correlates with Recurrence, Loss of Immune Infiltrate, and Chemoresistance. Clin. Cancer Res. 2019, 25, 1948–1956.
- Lahoz, S.; Rodríguez, A.; Fernández, L.; Gorría, T.; Moreno, R.; Esposito, F.; Oliveres, H.; Albiol, S.; Saurí, T.; Pesantez, D.; et al. Mutational Status of SMAD4 and FBXW7 Affects Clinical Outcome in TP53-Mutated Metastatic Colorectal Cancer. Cancers 2022, 14, 5921.
- Kawaguchi, Y.; Kopetz, S.; Newhook, T.E.; De Bellis, M.; Chun, Y.S.; Tzeng, C.-W.D.; Aloia, T.A.; Vauthey, J.-N. Mutation Status of RAS, TP53, and SMAD4 is Superior to Mutation Status of RAS Alone for Predicting Prognosis after Resection of Colorectal Liver Metastases. Clin. Cancer Res. 2019, 25, 5843–5851.
- Ansar, M.; Wang, C.-J.; Wang, Y.-H.; Shen, T.-H.; Hung, C.-S.; Chang, S.-C.; Lin, R.-K. SMAD3 Hypomethylation as a Biomarker for Early Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 7395.
- Shaker, O.G.; Mohammed, S.R.; Mohammed, A.M.; Mahmoud, Z. Impact of microRNA-375 and its target gene SMAD-7 polymorphism on susceptibility of colorectal cancer. J. Clin. Lab. Anal. 2018, 32, e22215.
- De Mattia, E.; Canzonieri, V.; Polesel, J.; Mezzalira, S.; Fratte, C.D.; Dreussi, E.; Roncato, R.; Bignucolo, A.; Innocente, R.; Belluco, C.; et al. SMAD3 Host and Tumor Profiling to Identify Locally Advanced Rectal Cancer Patients at High Risk of Poor Response to Neoadjuvant Chemoradiotherapy. Front. Pharmacol. 2021, 12, 778781.
- De Mattia, E.; Polesel, J.; Roncato, R.; Labriet, A.; Bignucolo, A.; Gagno, S.; Buonadonna, A.; D’Andrea, M.; Lévesque, E.; Jonker, D.; et al. IL15RA and SMAD3 Genetic Variants Predict Overall Survival in Metastatic Colorectal Cancer Patients Treated with FOLFIRI Therapy: A New Paradigm. Cancers 2021, 13, 1705.
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Exp. Cell Res. 2005, 307, 231–246.
- Maresca, C.; Di Maggio, G.; Stolfi, C.; Laudisi, F.; Colella, M.; Pacifico, T.; Di Grazia, A.; Di Fusco, D.; Congiu, D.; Guida, A.M.; et al. Smad7 Sustains Stat3 Expression and Signaling in Colon Cancer Cells. Cancers 2022, 14, 4993.
- Chen, W.; Wang, H.; Mi, S.; Shao, L.; Xu, Z.; Xue, M. ALKBH1-mediated m1 A demethylation of METTL3 mRNA promotes the metastasis of colorectal cancer by downregulating SMAD7 expression. Mol. Oncol. 2023, 17, 344–364.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
940
Revisions:
2 times
(View History)
Update Date:
06 Sep 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No